
Abstract
Abstract
Here, we report the isolation and characterization of embryonic stem (ES) cell-like cells from cloned blastocysts, generated using fibroblasts derived from an adult buffalo (BAF). These nuclear transfer embryonic stem cell-like cells (NT-ES) grew in well-defined and dome-shaped colonies. The expression pattern of pluripotency marker genes was similar in both NT-ES and in vitro fertilization (IVF) embryo-derived embryonic stem cell-like cells (F-ES). Upon spontaneous differentiation via embryoid body formation, cells of different morphology were observed, among which predominant were endodermal-like and epithelial-like cell types. The ES cell-like cells could be passaged only mechanically and did not form colonies when plated as single cell suspension at different concentrations. When F-ES cell-like, NT-ES cell-like, and BAF cells of same genotype were used for hand-made cloning (HMC), no significant difference (p > 0.05) was observed in cleavage and blastocyst rate. Following transfer of HMC embryos to synchronized recipients, pregnancies were established only with F-ES cell-like and BAF cell-derived embryos, and one live calf was born from F-ES cell-like cells. Further, when transfected NT-ES cell-like cells and BAF were used for HMC, no significant difference (p > 0.05) was observed between cleavage and blastocyst rate. In conclusion, here we report for the first time the derivation of ES cell-like cells from an adult buffalo, and its genetic modification. We also report the birth of a live cloned calf from buffalo ES cell-like cells.
Introduction
ES cell lines isolated from cloned blastocysts (NT-ES cell lines) have been established in mice (Kawase et al., 2000; Munsie et al., 2000; Wakayama et al., 2001), cattle (Wang et al., 2005) and nonhuman primates (Byrne et al., 2007). Human NT-ES cell line had been produced by transferring human somatic cells into enucleated rabbit oocytes (Chen et al., 2003). Although recent advances in induced pluripotent stem cells from farm animals (Roberts et al., 2009) may render unnecessary, the need for derivation of NT-ES cell lines, but for basic research they serve as excellent model, especially in mice. In case of farm animals, NT-ES cells can be derived from genetically elite individuals for use as donor cells in cloning, transgenesis (Wells et al., 2003) or for conservation if efficient protocols for cryopreservation of these cell lines can be developed.
Although the quantity and quality of yet unknown reprogramming factors present in oocyte determines the overall reprogramming efficiency during cloning, the degree of differentiation of donor cell also markedly influences cloning efficiency (Rideout et al., 2001). Compared to somatic cells, murine ES cells give higher cloning efficiency in terms of live offspring (Wakayama et al., 1999). No significant difference was observed in the cloning efficiency of mice when somatic and NT-ES cells derived from the same individual, were compared for cloning (Wakayama et al., 2005). However, an infertile mouse was successfully cloned using NT-ES cells as donor (Mizutani et al., 2008). Lanza et al. (2004) reported the use of NT-ES cells for restoring infracted myocardium in a mice model.
The use of ES cells has also been envisaged for increasing cloning efficiency in farm animals (Wells et al., 2003). ES cells have been used for reproductive cloning (Saito et al., 2003) and transgenesis (Cibelli et al., 1998) of cattle but when used to produce chimeric bovine offsprings they did not contribute to germline of the offspring (Iwasaki et al., 2000). The present study was thus carried out to (1) isolate and characterize NT-ES cell-like cells from hand-made cloned (HMC) buffalo blastocysts and study their differentiation potential, (2) compare the in vitro developmental potential of HMC embryos produced using buffalo NT-ES and adult fibroblast (BAF) cells of the same genotype and ES cell-like cells derived from IVF blastocysts (F-ES). Developmental potential of transgenic embryos generated using transfected NT-ES and BAF cells was also studied.
Materials and Methods
All chemicals and media were from Sigma Chemical Co. (St. Louis, MO, USA) and disposable plastic wares from Nunc (Roskilde, Denmark) unless otherwise mentioned. Animals trials carried out in this study were approved by the Animal Ethics Committee of the National Dairy Research Institute, Karnal, India.
IVF
Briefly, two straws of frozen–thawed ejaculated buffalo semen were washed twice with washing BO medium (BO medium containing 10 μgmL−1 heparin, 137.0 μgmL−1 sodium pyruvate and 1.942 μgmL−1 caffeine sodium benzoate). The pellet was resuspended in 0.5 mL of the washing BO medium. A and B grade cumulus–oocyte complexes (COCs) derived from slaughter house ovaries, and in vitro matured for 24 h as described previously (Chauhan et al., 1998), were washed thrice with the washing BO medium and transferred to 50 μL droplets (15–20 oocytes/droplet) of the capacitation and fertilization BO medium [washing BO medium containing 10 mgmL−1 fatty acid-free bovine serum alubmin (BSA)]. The spermatozoa in 50 μL of the capacitation and fertilization BO medium (≈3 million spermatozoa mL−1) were then added to the droplets containing the oocytes, covered with sterile mineral oil, and placed in a CO2 incubator (5% CO2 in air) at 38.5°C for 16–18 h. Embryo culture was performed in Research Vitro Cleave (K-RVCL-50, Cook®, Australia) medium with 50–60 presumed zygotes per well of a Nunc four-well dish.
Hand-made cloning
Primary cell culture of an adult Murrah buffalo (>6 years old) was established and prepared for HMC as reported earlier (Shah et al., 2008). Derivation of F-ES and NT-ES cell-like cells is explained below. For HMC, ES cell colonies (passage 10–15) were mechanically dissected and treated with trypsin for not more than 2 min to remove any traces of cells from the feeder layer. The colonies were then quickly transferred to T2 (where T denotes HEPES modified M-199 supplemented with 2.0 mM L-glutamine, 0.2 mM sodium pyruvate, 50 μgmL−1 gentamicin, and the following number denotes fetal bovine serum (FBS) in percent v/v) and single-cell suspension was made by vigorous pipetting. Cells were then transferred to the center of the four-well dish and used for HMC as donor cells. The recipient cytoplast maturation and processing was performed as described previously (Shah et al., 2008). Briefly, in vitro matured COCs with expanded cumulus were stripped off their cumulus investment and zona pellucida using hyaluronidase (0.5 mgmL−1 in T2) and pronase (2.0 mgmL−1 in T10), respectively. Oocytes with completely digested zona pellucida were transferred to T20 (T containing 20% FBS) and incubated at 38.5°C for 10–15 min or until a prominent protrusion cone was easily visible. Protrusion cone-guided bisection was performed using microblade (MicroBlades, MTB-05; Micromanipulator Microscope Company, Inc., Carson City, NV, USA) in 4 mL T20 with 2.5 μgmL−1 cytochalasin B. The larger demicytoplasts without protrusion cone were transferred to T20 and incubated for 10–15 min at 38.5°C to enable them to regain spherical shape. The enucleated demicytoplasts were immersed in phytohemagglutinin (0.5 mgmL−1 in T2) for 3–4 sec and transferred to T2 containing donor cells. Each demicytoplast was then allowed to attach to a single, rounded, medium-sized cell by gently rolling the demicytoplast over it. The couplets (demicytoplast–donor cell pairs) were transferred to fusion medium (0.3 M D-mannitol, 0.1 mM MgCl2, 0.05 mM CaCl2, and 1 mgmL−1 polyvinyl alcohol) for equilibration. A single-step fusion protocol was followed wherein a demicytoplast and a couplet were picked using a fine pulled capillary pipette (Unopette® Becton Dickinson, Franklin Lakes, NJ, USA) having an internal diameter of 100–150 μm. Initially, the couplet was expelled and aligned with an A.C. pulse (4 Volts) using BTX Electrocell Manipulator 200 (BTX, San Diego, CA, USA) so that the somatic cell faced the negative electrode, and immediately after alignment another demicytoplast was introduced into the fusion chamber (BTX microslide 0.5 mm gap, model 450; BTX, San Diego, CA, USA) close to the somatic cell. As soon as the somatic cell was sandwiched between the demicytoplasts, a single D.C. pulse (3.36 kVcm−1 for 4 μsec) was applied. The triplets were then incubated in T20 (for rounding up) for 6 h at 38.5°C. The reconstructed oocytes were activated by incubating in T20 containing 5 μM calcimycin A23187 for 5 min at 38.5°C. After washing thrice with T20, the oocytes were then incubated individually in 5 μL droplets of T20 containing 2 mM 6-dimethylamino purine, covered with mineral oil, and kept in incubator at 38.5°C for 4 h. The reconstructed, activated embryos were cultured in 400 μL of RVCL medium supplemented with 1% fatty acid-free (FAF) BSA in four-well dish (15–20 embryos per well), covered with mineral oil, and kept undisturbed in a CO2 incubator for 8 days. Cleavage was observed only after compaction stage at day 4 or 5. The total cell number of blastocysts were determined by staining them with Hoechst 33342 (50 μgmL−1) for 10 min and observing under an epifluorescence microscope.
ES cell derivation, characterization, and cryopreservation
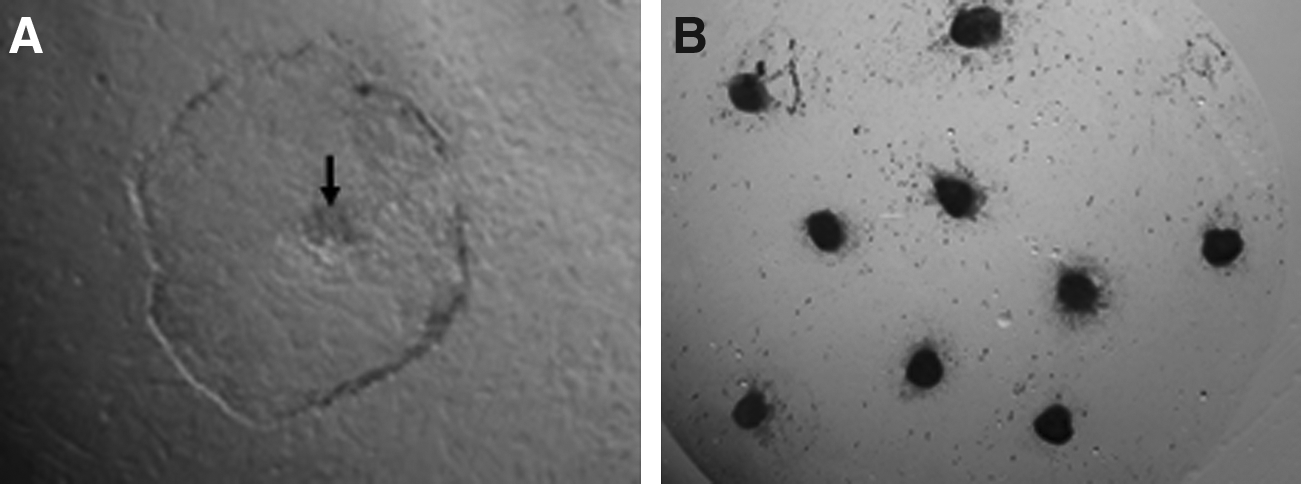
Inner cell masses (ICMs) were isolated mechanically from day 8 hatched blastocysts produced by IVF and HMC using microblade and were cultured on mitomycin C-inactivated (10 μgmL−1) buffalo fetal fibroblast feeder layers (Verma et al., 2007) in ES cell culture medium which comprised of Knockout-DMEM (Invitrogen Corporation, Carlsbad, CA, USA) + 15% Knockout serum replacer (Invitrogen) + 2 mM L-glutamine + 1% MEM nonessential amino acids + 0.1 mM β-mercaptoethanol + 1000 IUmL−1 LIF + 5 ngmL−1 bFGF-2 + 50 μgmL−1 gentamicin sulphate. Passage 1 was performed upon primary colony formation (Fig. 1A) and the medium was changed every 24 h. The colonies were passaged every 4–5 days either mechanically by bisecting them in 1:2 or 1:3 split ratio using a microblade, or by making a single-cell suspension by vigorous pipetting.
(
Expression of intracellular and surface markers, used for characterizing murine and human ES cells, was studied using reverse-transcriptase PCR (RT-PCR) and immunofluorescence (IF) staining as described earlier (Anand et al., 2009; Verma et al., 2007). The primer sequence and reaction conditions are given in Supplementary data (data are available online at http://www.liebertonline.com/cell; Supplementary Table 1). All primary antibodies (used at a dilution of 1:20), that is, anti-SSEA-1, SSEA-3, SSEA-4, TRA-1-60, and TRA-1-81 were purchased from Chemicon® International, Temecula, CA, USA, and FITC conjugated secondary antibodies (used at 1:200 dilution) were from Sigma Chemical Co. Alkaline phosphatase staining of ES cell colonies was performed using a kit (Catalog No. 86C) according to manufacturer's protocol (Sigma Chemical Co).
The ability of ES cell-like cells to give rise to the derivatives of the three germ layers was determined via embryoid body formation. Briefly, small, mechanically dissected clumps of ES cell-like cells, at different passages, were cultured in hanging drops (20 μL) for 48 h followed by suspension culture in Petri dishes. Derivatives of ectoderm, mesoderm, and endodermal origins in embryoid bodies were traced by studying the expression of transcriptional markers of the respective lineages by RT-PCR. Clonal origin of the NT-ES cell-like cells was confirmed by microsatellite analysis of DNA samples isolated from ES cell-like cells and blood of the adult animal, feeder layer DNA was used as negative control. Second round of HMC using NT-ES cells (passages 10–15) as donor was performed as described above. Both F-ES and NT-ES cells were monitored for the expression of pluripotency markers like alkaline phosphatase activity, SSEA-3, SSEA-4, OCT4, NANOG, and SOX-2 every fifth passage.
ES cell cryopreservation was performed as described earlier (Verma et al., 2007) with slight modifications. Colonies of ES cell-like cells, at different passages were vitrified in groups of four or five in French ministraws (IMV, L'Aigle, Cedex, France). Vitrification solutions were based on holding medium (HM, DMEM + 20% FBS). Colonies were incubated in vitrification solution 1 (VS 1; 10% dimethyl sulphoxide and 10% ethylene glycol) for 1 min, followed by incubation in VS2 (20% DMSO, 20% EG, and 0.5 M sucrose) for 20–30 sec and finally loaded into the straws. Just after sealing the open end of the straws with polyvinyl alcohol, these were immediately plunged into liquid nitrogen. For thawing, straws were plunged into water at 37°C for 30 sec; the sealed end was cut to allow colonies to enter and remain in the HM containing 0.2 M sucrose, for 1 min, followed by 5-min incubation in HM containing 0.1 M sucrose. The colonies were further incubated twice (5 min each) in HM before seeding on mitomycin C-treated feeder layer.
Transfection of BAF and ES cell-like like cells by nucleofection
BAF (passages 2–3) and NT-ES cell-like cells (passages 6–7) were transfected using Nucleofetor (Amaxa, Cologne, Germany) using the optimized program (U-23 and A-23, respectively), as suggested by the manufacturer. BAF were transfected as single-cell suspension and selected using G418 (800 μgmL−1), whereas, small clumps (10–15 cells) of NT-ES cells were transfected and selected using 100 μgmL−1 G418. Colonies showing homogenous and persistent GFP expression upon subsequent culturing were picked and propagated further. After confirmation of plasmid integration by PCR (Supplementary Fig. 1) the transfected NT-ES and BAF cell were used for HMC.
Synchronization of recipients and embryo transfer
Cycling buffaloes possessing a functional corpus luteum were treated with PGF2α analogue (Cloprostenol sodium, 500 μg) intramuscularly. Those exhibiting estrus at about 72 h after the treatment were selected as recipients. Day 8 HMC blastocysts were transferred nonsurgically to synchronized recipients after confirming the presence of corpus luteum. Pregnancies were confirmed by ultrasonography at days 40–50 after transfer. A single calf was delivered by Caesarean operation. Microsatellite analysis was carried out to confirm the origin of the cloned calf. The total genomic DNA from cloned calf, foster mother, and F-ES cell-like cells was isolated using standard phenol chloroform extraction method and used for amplification, using 13 primer pairs of microsatellite markers.
Statistical analysis
The data were analyzed using SYSTAT 7.0 (SPSS Inc., Chicago, IL, USA) and all values are presented as mean ± SEM unless indicated otherwise. Differences among means were analyzed by one-way ANOVA after arc-sine transformation of the percentage values. Differences were considered significant at p < 0.05.
Results
ES cell derivation and characterization
The rate of primary colony formation was similar (p > 0.05) for both F-ES and NT-ES groups (57.6 ± 8.8 vs. 33.2 ± 12.4, respectively). The time take for primary colony formation was highly variable in both the groups and varied from 5–10 days. ICMs attached and spread in almost similar pattern in both the groups. In the center of proliferated trophectodermal cell layer, a small dome-shaped clump of cells was visible (Fig. 1A), which was mechanically dissected out using a microblade, to discard the trophectoderm cells to the maximum extent possible. Clumps of ES cell-like cells were seeded individually in a 100 μL droplet containing ES cell culture medium on the feeder layer. On attachment and proliferation, cultures were designated as passage 1 and subsequently passaged in 1:2 or 1:3 split ratio, depending on size of the colony. The clonal origin of NT-ES cell-like cells was confirmed by microsatellite analysis.
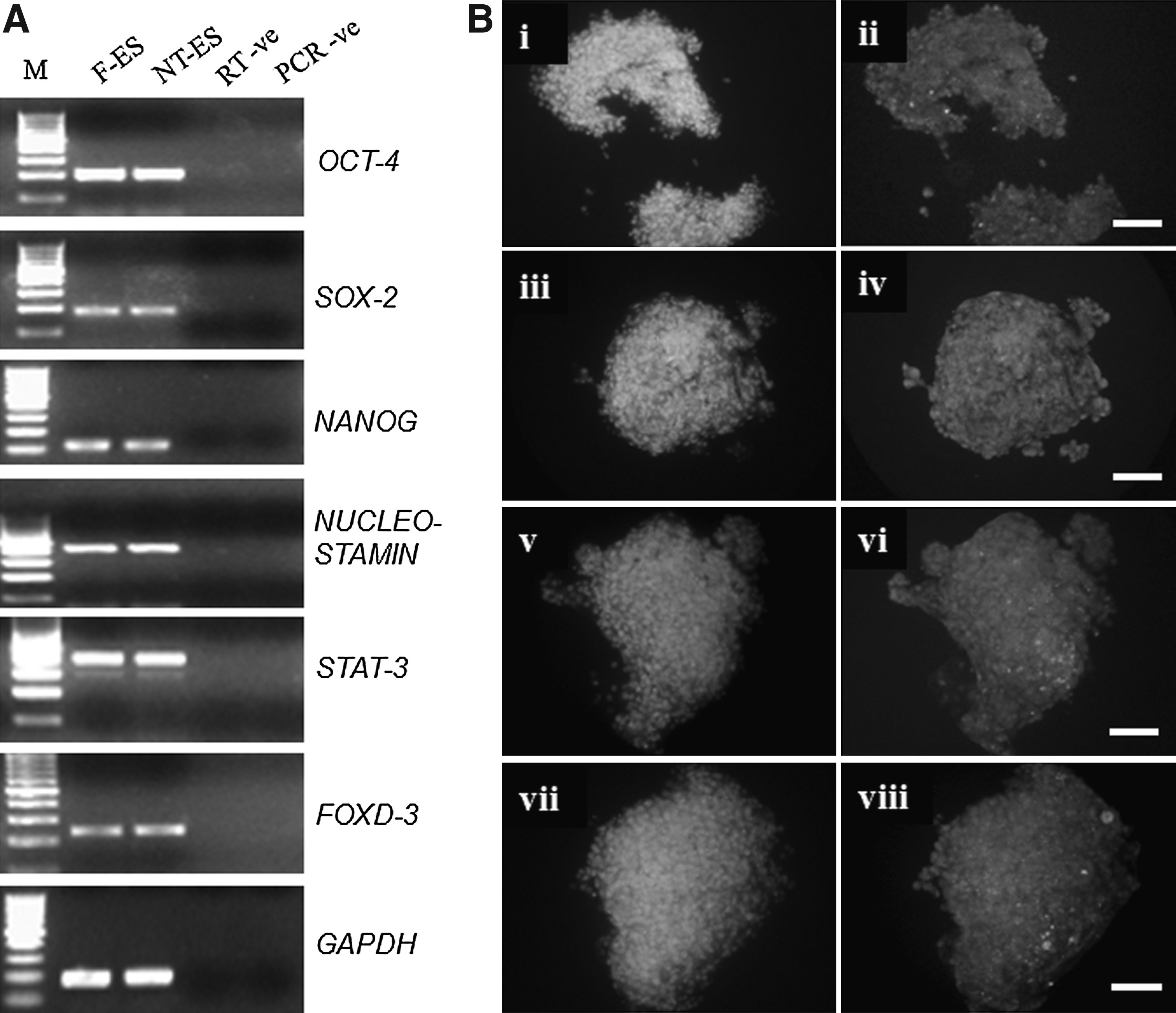
The expression pattern of pluripotency markers in both types of ES cells was similar (Fig. 2A). Both were positive for OCT-4, SOX-2, NANOG, NEUCLEOSTAMIN, STAT-3, and FOXD-3 as detected by RT-PCR. Using IF staining SSEA-3, SSEA-4, TRA-1-60, and TRA-1-81 were found to be expressed (Fig. 2B), whereas SSEA-1 was not expressed by both F-ES and NT-ES cell-like cells (Supplementary Fig. 2). NT-ES colonies exhibited high levels of ALP activity (Fig. 1B) as reported earlier for IVF-derived buffalo ES cell-like cells (Verma et al., 2007).
(
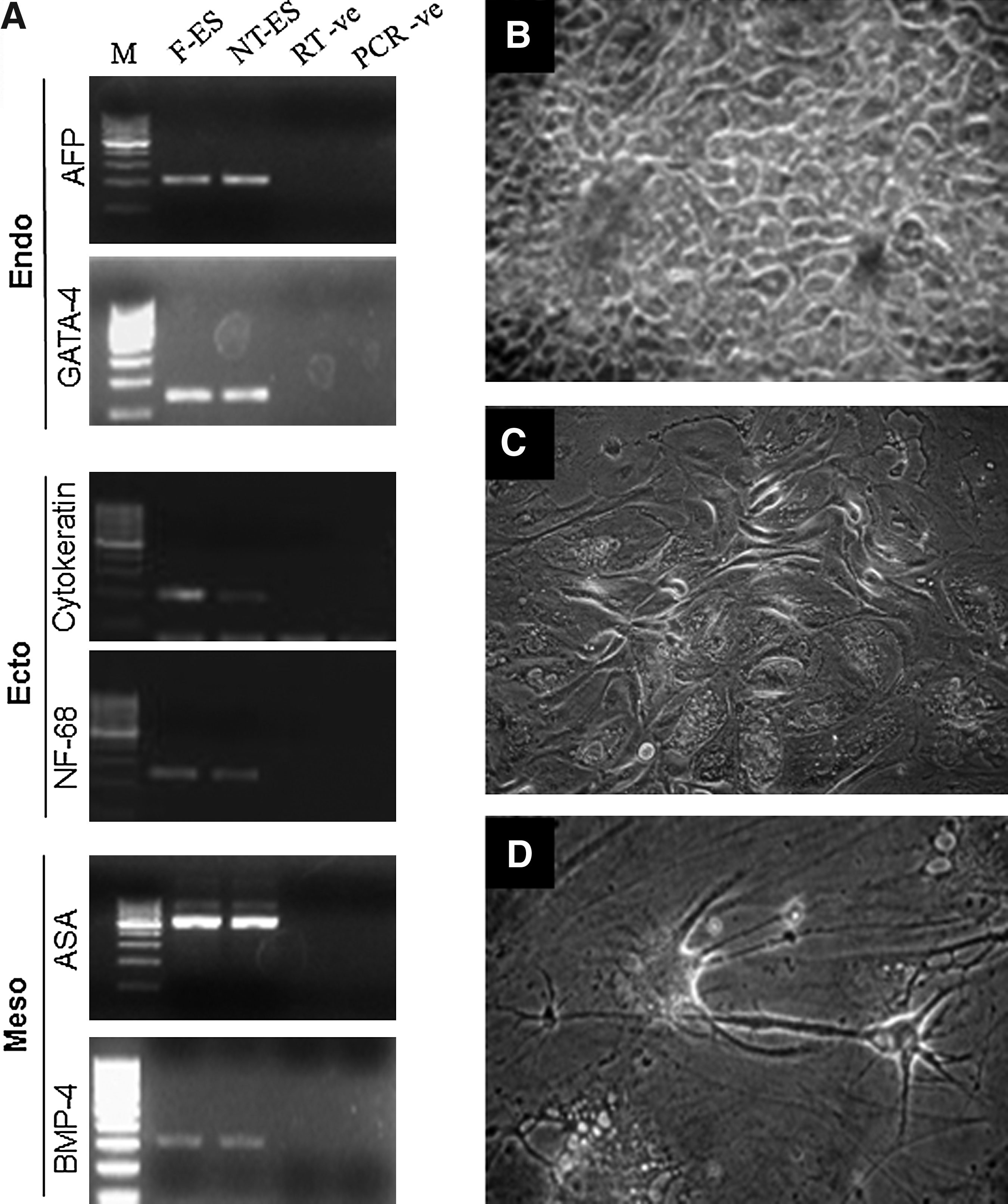
Expression of markers of the three germ layers (ectoderm, mesoderm, and endoderm) was found to be present in embryoid bodies produced from both NT-ES and F-ES cell-like cells (Supplementary Figs. 3A and B), as studied by RT-PCR (Fig. 3A). The cells in the colony outgrowths were found to spontaneously differentiate to cells resembling epithelial-like and fibroblast-like morphology, whereas neuron-like cells were found rarely (Fig. 3B–D).
(
Upon freeze–thaw, survival of both types of ES like cells was severely compromised. Although the colonies stained positive for ALP and other pluripotency markers, we were not able to maintain them for more than one to two passages, as none of the colonies could retain their normal developmental potential after cryopreservation.
Developmental competence of HMC embryos produced using NT-ES and BAF of same genotype and F-ES cells as nuclear donor
In the following experiment, donor cells of different degrees of differentiation derived from an adult buffalo were used for HMC (Table 1). The BAF (passages 10–15) derived from ear pinna and NT-ES cells (passage <15) derived from cloned blastocysts generated using these BAFs as donor cells, when used for HMC, gave almost similar cleavage rate (p > 0.05). The cleavage rate of F-ES cells was similar to that of the other groups. The blastocyst rate for BAF-derived HMC embryos (31.5 ± 5.6) was similar to that for those derived from NT-ES (28.6 ± 9.6) and F-ES cells (27.3 ± 4.0). The total cell number of the blastocysts (n = 20) produced using NT-ES (174.0 ± 5.7) and F-ES (180.7 ± 3.8) was significantly higher (p < 0.05) than that for blastocysts produced using BAF (157.0 ± 4.0). Transfer of HMC embryos (n = 14) reconstructed using F-ES cells to six recipients resulted in one pregnancy and a live female calf (Fig. 4) was born with a body weight of 32 kg, which is still surviving. No pregnancies resulted on transfer of HMC embryos (n = 12) reconstructed using NT-ES cells to four recipients. Two pregnancies were established when HMC embryos (n = 16) reconstructed using BAF were transferred to six recipients. However, these were found to be aborted at around day 90 of gestation. Microsatellite analysis revealed that the cloned calf was genetically similar to the respective donor cell. Genetically unrelated control DNA sample (surrogate buffalo) had multiple differences in analyzed microsatellite sequences (Table 2).

A cloned buffalo calf produced using buffalo ES cell-like cell as a donor.
Values with different superscripts within the same column differ significantly.
Values quoted as Mean% ± SEM.
The numbers indicate the size of each allele in base pairs.
Bold text indicates informative markers distinguishing genetically unrelated samples.
Production of transgenic buffalo embryos using transfected BAF and NT-ES cells
There was no significant difference in the growth characteristics between nontransfected and transfected BAF and NT-ES cell-like cells. But supplementation of G418 at 100 μgmL−1 was found to significantly reduce the proliferation of NT-ES cell-like cells (Supplementary Fig. 4). The colonies that expressed GFP consistently through subsequent passages were identified and propagated further. When transfected BAF (Fig. 5) and NT-ES cell-like cells (Fig. 6) were used as donor cells for HMC, no significant difference was observed in cleavage (70.4 ± 2.8 vs. 72.1 ± 3.5) and blastocyst rate (24.7 ± 2.0 vs. 23.2 ± 1.5). The total cell number of blastocysts (n = 20) generated using transfected NT-ES cell (162.4 ± 3.0) was significantly higher than that for blastocysts generated using transfected BAF cells (132.8 ± 2.4).

Buffalo fetal fibroblasts expressing GFP under bright-field (

A colony of transfected NT-ES cell-like cells expressing GFP under bright-field (
Discussion
The following study was carried out to derive ES cell-like cells from an adult buffalo whose productive and reproductive features are well characterized. A two-round NT approach was applied, wherein the first round NT provided the cloned blastocysts from which NT-ES cell-like cells were derived and used as nuclear donor for a second round of NT. BAF and NT-ES cell-like cells of the same genotype were transfected and were further used in HMC for production of cloned transgenic embryos.
The efficiency of primary colony formation from IVF and HMC blastocysts was found to be similar. The primary colony formation pattern and colony morphology in both the groups was identical. The colonies were compact, dome shaped, and had distinct boundary (Fig. 1A) as reported earlier (Verma et al., 2007; Wang et al., 2005). Although the colonies were not as compact compared to murine ES cell colonies, but still we had to vigorously pipette them for making single-cell suspension, treatment with trypsin alone was not sufficient to dissociate them into single cells. We also tried to passage these cells by making single-cell suspension but no colony formation occurred even at high-cell densities, suggesting a lower proliferation rate and/or higher incidence of apoptosis. Watanabe et al. (2007) reported that use of ROCK inhibitor (Y-27632) increases the clonal efficiency and survival after cryopreservation of human ES cells. The same might provide a solution to the problem of low proliferation and survival rate after cryopreservation of ES cell-like cells derived from farm animals.
In cattle, whereas ES cell-like cells colonies have been reported to be positive for ALP staining in some reports, it was not detected in others (Cibelli et al., 1998; Mitalipova et al., 2001; Stice et al., 1996). However, in the present study, both F-ES and NT-ES cell colonies stained strong positive for ALP and expressed the commonly used markers for pluripotency, that is, OCT-4, NANOG, and SOX-2 as detected by RT-PCR. We also found the expression of some other reported markers of pluripotency like FOXD-3, NEUCLEOSTAMIN, and STAT-3 in these colonies. By using IF staining we were able to detect the expression of SSEA-4, SSEA-3, TRA-1-60, and TRA-1-81, but not of SSEA-1. Several stage-specific embryonic antigens (SSEAs) such as SSEA-1, SSEA-3, and SSEA-4, which show marked developmental regulation in mouse and human embryos are used as markers to monitor pluripotency of ES cells in these species along with other surface markers like tumor rejection antigen (TRA)-1-60 and TRA-1-81 (Henderson et al. 2002). The transcription based markers like OCT-4, NANOG, SOX-2, NUCLEOSTAMIN, STAT-3, and FoxD3, which are used for the characterization of pluripotent stem cells across several different species, play a critical role in maintaining the pluripotency of ES cells. The factors are expressed in higher quantities in pluripotent ES cells and are downregulated during differentiation. Huang et al. (2010) reported the expression of SSEA-1, SSEA-3, and SSEA-4 in IVF-derived buffalo ES cells, whereas parthenogenetically derived ES cells exhibited SSEA-1 and SSEA-4, TRA-1-60 and TRA-1-81, but not SSEA-3 (Sritanaudomchai et al., 2007). The reason for these variations is still not clear, but more conclusive results can be drawn if bovine or bubaline-specific antibodies can be developed and used for this purpose.
Previous research in mice had shown that NT-ES cell lines are characteristically and functionally equivalent to fertilization-derived ES cell lines. Transcriptional profiling of murine NT-ES cells and IVF-derived ES cells had revealed similarities between the two (Brambrink et al., 2006). MicroRNA and proteomic profiling of the two cell types had also revealed similar profiles (Ding et al., 2009). Both NT-ES and F-ES cell lines exhibited similar developmental potency in terms of chimeric offspring generation using tetraploid embryo complementation technique (Brambrink et al., 2006).
Many somatic cell types, including mammary epithelial cells, ovarian cumulus cells, fibroblast cells from skin and internal organs, various internal organ cells, granulosa, myoblast, neurons, Sertoli cells, macrophage, blood leukocytes, and ES cells have been successfully utilized for nuclear transfer. To date, no particular cell type has presented an overwhelming advantage over the others. However, the cloning efficiency with blastomeres or ES cells has been observed to be higher than that with somatic or terminally differentiated cells, probably because of the ease of reprogramming of these uncommitted cell types (Oback 2009). Age and degree of differentiation (Hochedlinger and Jaenisch, 2002; Oback and Wells, 2002; Tian et al., 2003) of donor cell significantly influences cloning efficiency. When ES cells were used as nuclear donor, lower percentages were obtained in terms of morula and blastocyst rate, but live offspring birth rate was higher compared to when cumulus cells were used as donor cells (Wakayama et al., 1999). The possibility of using ES cell-like cells, either IVF- or NT-derived, as donors, was explored in the present study in view of the very low overall cloning efficiency obtained with somatic cells in buffalo (Shah et al., 2009). Also, it would be interesting to compare the cloning efficiency obtained with F-ES and NT-ES cells because such information is not available in any farm animal species. In our study, the percentage of cleaved embryos in F-ES, NT-ES, and BAF groups did not differ significantly, which suggests that the ES cell-like cells were similar to somatic cells in terms of cell cycle. We used F-ES cell-like cells to determine whether there is some detrimental effect of the first round of HMC for derivation NT-ES cells on the developmental competence of embryos. We found no significant difference in the developmental competence of embryos generated using F-ES and NT-ES cell-like cells. In mice, when neural stem cells were used for NT, the cleavage rate was similar to those of cumulus cells and differentiated neural stem cell-derived cloned embryos (Mizutani et al., 2006). However, in another study, the cleavage rate for ES cell-derived cloned embryos was significantly lower, which was attributed to their higher proliferation rate (Wakayama et al., 1999). In bovines, though, a direct comparative study was not performed, but ES cells, when used for NT, provided cleavage rate similar to those obtained with somatic cells, but the blastocyst rate was very low, which was attributed to lack of coordination of cell cycle between the ES cells and oocyte activation (Saito et al., 2003). In mice, somatic and NT-ES cells derived from the same individuals gave similar rate in terms of live offspring born, when used for producing cloned mice, although marked differences were reported among NT-ES cell lines of different genetic backgrounds to produce cloned offspring via NT (Wakayama et al., 2005). Until now, ES cells capable of generating germ line chimeras have not been obtained in farm animals, therefore, the pluripotent nature of these cells is not well defined and based only on expression of pluripotency markers defined for mice and human ES cells, which could be misleading (Munoz et al., 2008).
We also used transfected BAF and NT-ES cell-like cells of same genotype for use as donor cells in HMC and no significant difference was observed in cleavage and blastocyst rate in the two groups. In our earlier study, we have found that transfection and subsequent selection significantly influences the developmental potential of HMC embryos (unpublished data). Because we had obtained best results using nucleofection, which had resulted in highest transfection efficiency and acceptable survival rate in our earlier study, it was used for transfection in the present study. There was no detrimental effect of the transfection procedure on NT-ES cells in comparison to BAF but NT-ES cells were found to be highly sensitive to G418 selection (Supplementary Fig. 4). No significant difference was observed between transfected and non transfected NT-ES cell colonies in terms of growth, proliferation, and embryoid body formation (Supplementary Fig. 3B and C).
In contrast to NT-ES cell lines of mice, cloned embryos, and fetus derived by NT exhibit marked abnormalities in gene expression pattern. Bortvin et al. (2003) reported incomplete reactivation of Oct-4-related genes in cumulus cell-derived cloned mice embryos, whereas ES cell-derived cloned embryos faithfully expressed these genes. Retention of epigenetic memory by the differentiated donor cells has been attributed to aberrant gene expression in NT-derived embryos and clones (Jaenisch 2004; Ng and Gurdon 2005). It has been proposed that during derivation of NT-ES cell lines, the epigenetic memory is erased and a pattern similar to that of F-ES cells is reset. As such, the derivation of NT-ES cell lines is much easier compared to production of live cloned offspring (Wakayama et al., 2006). Although there is no direct evidence that cloning related abnormalities can be abrogated by the use of ES cells, the phenomenon of epigenetic reprogramming can be made more efficient by the use of this cell type.
In conclusion, we have demonstrated the derivation of NT-ES cell-like cells from an adult animal, which exhibit pluripotency marker expression and differentiation pattern comparable to that of F-ES like cells. Furthermore, their capacity to support in vitro development upon nuclear transfer was similar to those of genotype matched BAF and unrelated F-ES like cells. A single live surviving cloned calf was produced from F-ES cell-like cells. Genetic modification of NT-ES and BAF cells was carried out and used to produce transgenic HMC blastocysts.
Footnotes
Acknowledgments
The authors thank Dr. Anandlaxmi for providing access to Amaxa Nucleofector (Animal Physiology Division, NDRI) and Dr. M.S. Tantiya (National Bureau of Animal Genetic Resources) for help with microsatellite analysis. The present work was funded by National Agriculture Innovative Project (NAIP) Grant to M.S.C. (C-2067 and 075) and S.K.S. (C 2-1-(5)/2007).
Author Disclosure Statement
The authors declare that no conflicting financial interests exist.
References
Supplementary Material
Please find the following supplemental material available below.
For Open Access articles published under a Creative Commons License, all supplemental material carries the same license as the article it is associated with.
For non-Open Access articles published, all supplemental material carries a non-exclusive license, and permission requests for re-use of supplemental material or any part of supplemental material shall be sent directly to the copyright owner as specified in the copyright notice associated with the article.