
Abstract
Abstract
The removal of chromosomes from recipient oocytes is one of the key steps in nuclear transfer cloning. Although microtubule interrupters have been successfully used for oocyte enucleation, their potential side effect on oocyte developmental potential should be considered, and less harmful drugs should be explored for chemical-assisted enucleation. Based on our previous findings that any maturation promoting factor-activating agent induces ooplasmic protrusion without disrupting microtubules, we have studied the feasibility to use caffeine or MG132 for chemical-assisted enucleation. Experiments using goat oocytes showed that treatments for 30 min with 1-mM caffeine or 5-μM MG132-induced ooplasmic protrusions in about 85% of the oocytes, a percentage similar to that achieved with optimal demecolcine treatment. Rates of enucleation, cell fusion and in vitro blastulation were similar among caffeine, MG132, and demecolcine enucleation but significantly higher than blind aspiration. Furthermore, neither rates of pregnancy on days 90 and 120 nor the general rate of live births/embryos transferred differed significantly (p > 0.05) between caffeine and demecolcine enucleation. Although oocytes treated with caffeine did not retract protrusions until 2 h, many oocytes treated with MG132 withdrew protrusions as early as 0.5 h after treatment. The optimal treatment to induce ooplasmic protrusion in 75% pig oocytes was 8-mM caffeine for 60 min. Mouse oocytes responded poorly to demecolcine or caffeine with less than 40% forming inconspicuous protrusions following optimal treatments. It is concluded that caffeine can be used for enucleation of goat and pig oocytes with similar results as demecolcine, and live kids were born after caffeine-assisted enucleation.
Introduction
The removal of chromosomes from recipient oocytes is one of the key steps in NT cloning. Because the chromosomes of mammalian oocytes are difficult to observe under a light microscope, they are usually removed by blindly aspirating a large volume of cytoplasm underneath the first polar body (Li et al., 2004; Tsunoda and Kato, 2000). Because it has been found that the position of the first polar body relative to the M II spindle changed soon after oocyte maturation (Miao et al., 2004), and that the ooplasm volume removed during enucleation must be tightly controlled to increase the developmental potential of reconstituted embryos (Zakhartchenko et al., 1997; Zhou and Tan, 1996), an alternative should be found to the blind aspiration method of oocyte enucleation. Although chromosomes could be located and removed with a better control of ooplasmic volume after Hoechst staining, the exposure to ultraviolet irradiation may cause damage to cytoplasmic organelles and mitochondrial DNA, resulting in compromised cloning efficiency (Dominko et al., 2000). Chemically assisted enucleation seems to be an approach of choice with a better control of the ooplasmic volume removed and avoidance of exposure to ultraviolet irradiation. Microtubule disrupters such as demecolcine and nocodazole have been used for chemical-assisted oocyte enucleation in pigs (Kawakami et al., 2003; Li et al., 2006; Yin et al., 2002), cattle (Tani et al., 2006; Vajta et al., 2005), goats (Lan et al., 2008), rats (Hayes et al., 2001), and mice (Costa-Borges et al., 2009). However, although it is known that demecolcine inhibits mitosis at metaphase by inhibiting spindle formation during somatic cell division (Sutton, 1965), the mechanism for chemically assisted enucleation of oocytes needs further investigation. Furthermore, enucleation with both demecolcine and nocodazole disrupted chromosome spindles (Costa-Borges et al., 2009; Lan et al., 2008), and long-term demecolcine treatment was found detrimental to the developmental potential of mouse embryos (Kato and Tsunoda, 1992) and rat oocytes (Galat et al., 2007). Therefore, the potential side effect of microtubule disruptors should be considered and alternative, less harmful methods must be developed for chemical-assisted enucleation.
Our previous study using goat oocytes indicated that demecolcine induced ooplasmic protrusion by increasing maturation promoting factor (MPF) and mitogen activated protein kinase (MAPK) activities via activating MAD2 (Wu et al., 2010). Both caffeine and MG132 are MPF-activating agents; whereas MG132, a specific inhibitor of proteasome catalytic activity, activates MPF by preventing cyclin B degradation (Glotzer et al., 1991; Josefsberg et al., 2000), caffeine increases MPF activity by inhibiting Myt1/Wee1 and dephosphorylating CDC2A (Kikuchi et al., 2000; Smythe and Newport, 1992). We therefore proposed the possibility to use caffeine or MG132 as a less harmful drug to chemically assist oocyte enucleation. This proposal was tested in this study. The protocol for caffeine- or MG132-assisted enucleation of goat oocytes was first optimized, and the efficiency of enucleation, cell fusion, and embryo development both in vitro and in vivo were then compared between caffeine-, MG132-, and demecolcine-assisted enucleation. The feasibility to use caffeine for oocyte enucleation in other animals was finally tested using pig and mouse oocytes. The results showed that caffeine could be used for enucleation of goat and pig oocytes with similar results as demecolcine, and live kids were born from goat embryos reconstructed following caffeine-assisted enucleation.
Materials and Methods
Chemicals were purchased from Sigma Chemical Co (St. Louis, MO, USA) unless otherwise specified.
Collection and in vitro maturation of goat and pig oocytes
Caprine and porcine ovaries were obtained from a local abattoir, and transported within 3 h to the laboratory in sterilized saline containing 100 IU/mL penicillin and 0.05 mg/mL streptomycin and maintained at 30–35°C. Oocyte aspiration and selection were performed in the handling medium: Dulbecco's phosphate-buffered saline (D-PBS) supplemented with 0.1% of polyvinyl alcohol (PVA). Cumulus–oocyte complexes (COCs) were aspirated with a syringe from 1.5–4 mm goat or 3–6 mm pig follicles. The COCs were examined under a stereomicroscope, and only those with more than three complete layers of cumulus cells and a finely granulated homogeneous ooplasm were selected for maturation culture. The culture medium was TCM-199 (Gibco, Grand Island, NY, USA) supplemented with 10% (v/v) FCS (Gibco), 1 μg/mL 17 β-estradiol, 24.2 mg/L sodium pyruvate, 0.05 IU/mL FSH, 0.05 IU/mL LH, 10 ng/mL EGF, and 100 μM cysteamine and 200 μM cystine. Goat and pig COCs were cultured for 19 and 44 h, respectively, in groups of around 20 in droplets of 100 μL, covered with mineral oil, at 38.5°C in 5% CO2 in humidified air.
Recovery of mouse oocytes
Mice of the Kunming breed were kept in a room with 14 h/10 h light–dark cycles, the dark starting form 8 p.m. The animals were handled by the rules stipulated by the Animal Care and Use Committee of Shandong Agricultural University. Female mice, 6–8 weeks after birth, were induced to superovulate with equine chorionic gonadotropin (eCG, 10 IU, i.p.) followed 48 h later by human chorionic gonadotropin (hCG, 10 IU, i.p.). Both eCG and hCG used in this study were from Ningbo Hormone Product Co., Ltd (P. R. China). The superovulated mice were sacrificed 13 h after hCG injection and the oviductal ampullae were broken in the handling medium to release COCs.
Oocyte enucleation
Blind aspiration
Freshly matured goat oocytes were stripped of their cumulus cells and those with intact first polar bodies but without spontaneous ooplasmic protrusion were micromanipulated in the handling medium for enucleation. The first polar body and 1/5–1/4 of the cytoplasm underneath were removed using a glass micropipette with 20 μm of inner diameter.
Chemical-assisted enucleation
Freshly matured oocytes were denuded of cumulus cells and those with intact first polar bodies but without spontaneous protrusion were incubated at 38.5 (goat and pig) or 37.5°C (mouse) in mD-PBS containing different concentrations of demecolcine, caffeine, or MG132 for different times according to the experimental design. At the end of incubation, oocytes were examined under a microscope for the formation of cytoplasmic protrusions and were enucleated by aspirating the protrusions with an enucleating pipette when necessary. Enucleation efficiency for both blind aspiration and chemical-assisted enucleation was evaluated under a fluorescence microscope after the manipulated oocytes were stained with 10 μg/mL Hoechst 33342 contained in mD-PBS.
Preparation of donor cells
Goat cumulus cells were collected through hyaluronidase digestion of the cumuli of in vitro matured goat oocytes. After centrifugation, the cumulus cells were seeded and cultured in wells of a 96-well plate containing DMEM/F12 + 10% FCS at 38.5°C in 5% CO2 in air. When confluence was achieved, cells were trypsinised for 3 min, and the recovered cells were centrifuged and the resulting pellets were resuspended in the above medium and subcultured until use. The synchronization of the cell cycle at the G0/G1 stage was achieved by cell contact inhibition for 3–5 days. Transgenic goat fetal fibroblast cells with lactoferrin or Fat1 gene were prepared and provided frozen by Professor Wei Shen at Qingdao Agricultural University, P.R. China. The frozen cells were thawed and cultured in the same way as for cumulus cells before NT.
Nuclear transfer
A micropipette containing the donor cell was introduced through the slit of zona made during enucleation, and the cell was inserted between the zona and the cytoplast membrane to facilitate close membrane contact and subsequent fusion. The reconstructed embryos were electrically fused at 23–24 h of in vitro maturation in fusion medium comprised of 0.3 M mannitol, 0.05 mM CaCl2, 0.1 mM MgSO4, and 0.1% bovine serum albumin (BSA). The reconstructed embryos were manually aligned so that the contacting membranes of the cytoplast and donor cell were parallel to the electrodes. Fusion was induced with a single DC pulse of 1.2 kV/cm for 40 μsec.
Activation
The fused embryos were cultured in the CR1aa medium (Rosenkrans et al., 1993) supplemented with 3 mg/mL BSA and 5% fetal calf serum (FCS) for 30 min before activation. The ionomycin and 6-DMAP stocks were prepared in dimethyl sulfoxide (DMSO) and diluted to the desired concentrations in CR1aa supplemented with 3 mg/mL BSA and 5% FCS before use. The NT embryos were first exposed to 2.5 μM ionomycin for 1 min at room temperature, and then were incubated at 38.5°C under 5% CO2 in humidified air in CR1aa containing 2 mM 6-DMAP for 2 h.
Embryo culture
The NT and parthenogenetic embryos were cocultured on cumulus cell monolayer in CR1aa containing 3 mg/mL BSA and 5% FCS. To prepare cumulus cell monolayer, cumulus cells were collected from in vitro matured COCs and cultured in DMEM/F12 supplemented with 10% FCS in wells of a 96-well culture plate. The DMEM/F12 in wells with growing monolayer were replaced with CR1aa and equilibrated for 12 h prior to coculture. After activation treatments, embryos were transferred to the wells with cumulus cell monolayer and incubated at 38.5°C under 5% CO2 in humidified air. Cleavage and blastocyst formation was examined at 24 h and on day 9 of culture, respectively.
Embryo transfer
Lubei white recipient does were selected from those exhibiting a natural estrus 1 day prior to scheduled embryo transfer. The NT embryos that had been cultured for 16–20 h after fusion were transferred into oviducts of the recipients at 24–36 h after estrus. Pregnancy of the recipients was determined by ultrasound examination at different times following embryo transfer.
Data analysis
In all experiments but the one for in vivo development of cloned embryos, there were at least three replicates for each treatment. Data were arc sine transformed and analyzed with ANOVA; a Duncan multiple comparison test was used to locate differences. In the experiment for in vivo development of cloned embryos, independent samples t-tests were conducted to compare the effects of demecolcine and caffeine enucleation. The soft ware used was SPSS (Statistics Package for Social Science). Data are expressed as mean ± SE, and p < 0.05 was considered significant.
Results
Effects of concentration and duration of caffeine and MG132 treatment on the formation of ooplasmic protrusions in goat oocytes
Freshly matured goat oocytes with intact first polar bodies but without spontaneous protrusion were treated for different times with caffeine or MG132 at different concentrations before examination for ooplasmic protrusion. After treatment with caffeine or MG132, goat oocytes formed cytoplasmic protrusions that contained a condensed chromosome mass (Fig. 1A and A′). When oocytes were treated for 30 min with different concentrations of caffeine or MG132, satisfactory percentages of oocytes forming cytoplasmic protrusions were obtained at 1 mM caffeine or 5 μM MG132, and did not increase significantly when drug concentration was increased further (Table 1). When oocytes were incubated with 1 mM caffeine or 5 μM MG132 for different times, significantly more oocytes formed protrusions when incubated for 30 or 60 min than incubated for 15 min. Therefore, all the oocytes were treated with 1 mM caffeine or 5 μM MG132 for 30 min in the following experiments.
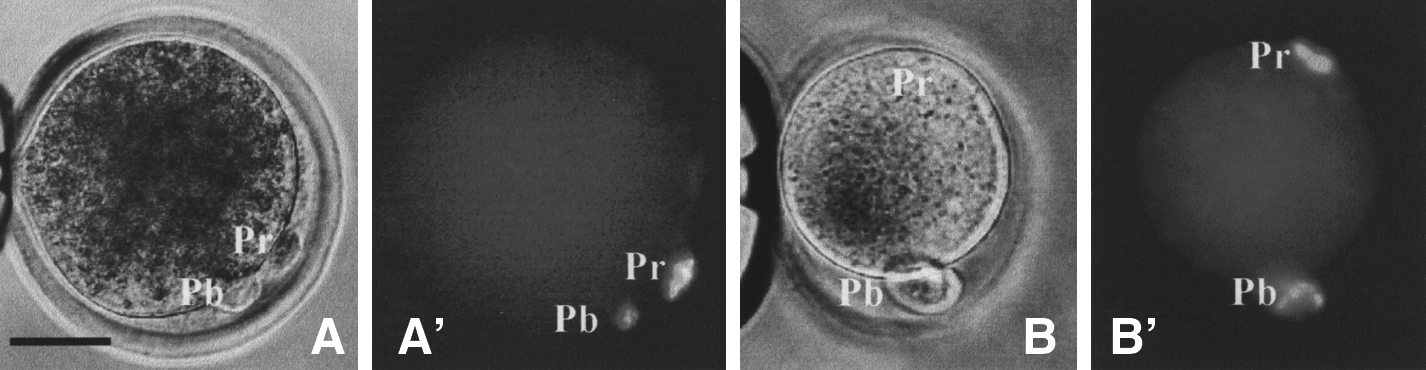
Micrographs of goat (
Values without a common letter in their superscripts in the same column differ (p < 0.05).
Effects of chemical treatment for enucleation on parthenogenetic development of goat oocytes
To test the detrimental effect of chemical treatment for enucleation on oocytes, goat oocytes were first cultured for 30 min with or without demecolcine, caffeine, or MG132 and then cultured for 4 h in maturation medium before activation treatment with ionomycin plus 6-DMAP. Oocytes after activation treatment were cultured for parthenogenetic development. Neither cleavage nor blastocyst formation percentages of oocytes treated with demecolcine, caffeine, or MG132 differed from those of oocytes activated without chemical treatment (Fig. 2). The results suggested that caffeine or MG132 treatment had no detrimental effect on oocyte developmental potential.

In vitro development of goat oocytes after parthenogenetic activation following treatment with demecolcine, caffeine, or MG132 to induce protrusions. Freshly matured oocytes were activated without protrusion-inducing treatment to serve as controls. Each treatment was repeated three times and each replicate contained 20–25 oocytes. a: Values without a common letter above their bars differ within two-cell and blastocyst groups (p < 0.05).
Enucleation efficiency, cell fusion, and development of cloned embryos after chemical-assisted or blind aspiration enucleation
Oocytes that had been enucleated by either chemical-assisted or blind aspiration enucleation were electrically fused with cumulus cells that were synchronized at G0/G1 stages. Fused embryos were cocultured on cumulus cell monolayer after activation treatment. Some of the cultured embryos were transferred to recipient does for in vivo development at 16–20 h of culture while others were cultured for 9 days for in vitro development. The results showed that enucleation rates were significantly higher after chemical-assisted enucleation than after blind aspiration (Table 2). Although rates of cell fusion were similar, rates of blastocysts were significantly higher after chemical enucleation than after blind aspiration. Rates of pregnancy on day 90 and day 120 and the general rate of live births calculated from embryos transferred did not differ significantly between caffeine and demecolcine enucleation, either within donor cell groups or in total of all the three donor cell types (Table 3). Together, the data indicated that embryos derived from caffeine-assisted enucleation had a similar developmental potential as those from demecolcine enucleation.
Values without a common letter in their superscripts in the same column differ (p < 0.05).
Independent samples t-tests were conducted using SPSS to compare the effects of demecolcine and caffeine enucleation, and results showed that rates of pregnancy on day 90 and day 120 and the general rate of live births calculated from embryos transferred did not differ significantly between caffeine and demecolcine enucleation, either within donor cell groups or in total of all the three donor cell groups.
CC, cumulus cells; LF, lactoferrin transgenic fetal fibroblast cells; FF, Fat1 transgenic fetal fibroblast cells.
Persistence of the cytoplasmic protrusions induced with demecolcine, caffeine, or MG132
To determine the time that the chemical-induced ooplasmic protrusions persisted, goat oocytes that had formed protrusions following different treatments were placed in mD-PBS and checked at different time points for the disappearance of protrusions. The results showed that although oocytes treated with demecolcine or caffeine did not retract their protrusions until 2 h, many of the oocytes with MG132-induced protrusions did as early as 0.5 h after they were placed in mD-PBS (Fig. 3). Thus, MG132 should be excluded as an appropriate drug for enucleation due to the limited persistence of protrusions.
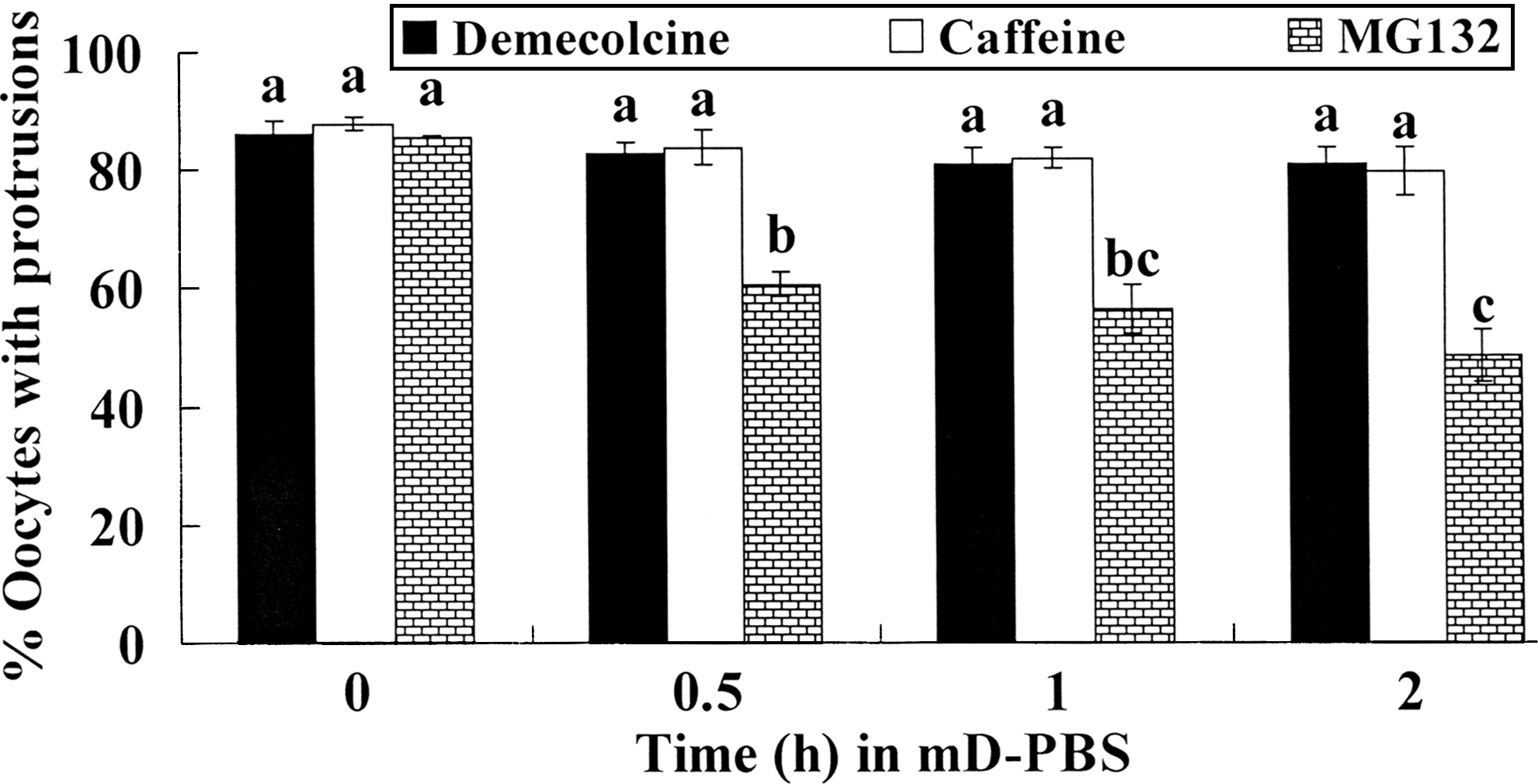
Persistence of the cytoplasmic protrusions induced with demecolcine, caffeine, or MG132 after goat oocytes were placed in mD-PBS for different times. In each treatment, 50 to 60 oocytes were observed. a–c: Values without a common letter above their bars differ (p < 0.05).
Caffeine induced ooplasmic protrusion efficiently in pig oocytes but not as efficiently in mouse oocytes
To see whether caffeine can induce ooplasmic protrusion in other species as well, freshly matured pig and mouse oocytes were treated for different times with demecolcine or caffeine of different concentrations. Percentages of pig oocytes forming protrusions increased with both concentration and duration of caffeine treatment but did not reach the highest until treatment with 8 mM caffeine for 60 min (Table 4). This suggested that pig oocytes were less sensitive than goat oocytes to chemical treatment to induce ooplasmic protrusion. The mouse oocytes responded poorly to both demecolcine and caffeine with less than 40% forming inconspicuous protrusions (Fig. 1B and B′) following the optimal treatments with 0.4 μg/mL demecolcine or 2 mM caffeine for 15 or 30 min (Table 5). Treatments for 30 min with 1 μg/mL demecolcine or 4 mM caffeine failed to increase percentages of mouse oocytes forming protrusions (data not shown).
Values without a common letter in their superscripts in the same column differ (p < 0.05).
Values without a common letter in their superscripts differ (p < 0.05).
Discussion
The present results indicated that although treatment with 1 mM caffeine for 30 min induced ooplasmic protrusion in about 85% of goat oocytes, the maximum protrusion formation in pig oocytes was not achieved until caffeine treatment increased to 8 mM for 60 min. The optimal demecolcine treatment was 0.8 ng/mL for 30 min to induce ooplasmic protrusions in over 90% of the goat oocytes (Lan et al., 2008). This optimal concentration of demecolcine for goat oocytes was almost 500-fold lower than that (0.4 to 0.5 μg/mL) used for porcine (Kawakami et al., 2003; Li et al., 2006; Yin et al., 2002) and bovine oocytes (Tani et al., 2006; Vajta et al., 2005). Although the reason for this tremendous difference in demecolcine and caffeine concentration and/or duration between species is unknown, it does suggest that protocols to induce ooplasmic protrusion must be optimized for different species, as long-term demecolcine treatment was found detrimental to the developmental potential of mouse embryos (Kato and Tsunoda, 1992) and rat oocytes (Galat et al., 2007). In addition, oocytes forming protrusions after drug treatment were usually placed in medium containing cytochalasin B to remove mechanically the maternal chromosomes (Yin et al., 2002). This practice needs reconsideration because our previous results showed that cytochalasin B promoted retraction of cytoplasmic protrusions (Lan et al., 2008). As 100% of the goat oocytes survived removal of caffeine-induced protrusions in the absence of cytochalasin B and a reasonable proportion of them developed normally after NT in this study, we recommend ooplasmic protrusions be removed without cytochalasin B treatment in this species.
In this study, rates of enucleation, cell fusion, and blastulation were similar among caffeine, MG132, and demecolcine enucleation but were significantly higher than those achieved by blind aspiration were. Embryo transfer experiments showed that both rates of pregnancy on days 90 and 120 and the general rate of live births/embryos transferred were similar between caffeine and demecolcine enucleation. This suggests that embryos derived from caffeine-assisted enucleation had a similar developmental potential as those produced by demecolcine enucleation. The efficiency of mammalian cloning from somatic cells is extremely low, with most clones failing after implantation (Hill et al., 1999; Lan et al., 2006; Renard et al., 1999; Sakai et al., 2005; Zakhartchenko et al., 1999). Thus, blastocyst development is only one step along the road to the production of a live offspring (Lonergan et al., 2003). In fact, blastocysts produced in vitro or by nuclear transfer are not homogenous in developmental potential; they have been found different in gene expression (Boiani et al., 2005; Eckardt and McLaughlin, 2004) and apoptotic status (Lonergan et al., 2003; Melka et al., 2009), for example. Although the better preimplantation development of embryos cloned by chemical-assisted enucleation compared to blind aspiration could be attributed to reduced removal of ooplasm (Peura et al., 1998; Zakhartchenko et al., 1997; Zhou and Tan, 1996). The difference between caffeine- and demecolcine-assisted enucleation was that demecolcine disrupted spindles while caffeine did not. As a result, although microtubules were completely removed together with the chromosome spindle after caffeine treatment, some microtubules might be left in the recipient cytoplast following demecolcine enucleation. What the redundant microtubules would do to the reconstructed embryos after nuclear transfer is not clear but warrants an investigation.
The present results indicated that while oocytes treated with demecolcine or caffeine did not retract protrusions until 2 h, many oocytes treated with MG132 withdrew their protrusions as early as 0.5 h after treatment. Our previous study in goat oocytes (Wu et al., 2010) demonstrated that demecolcine activates MPF by disassembling spindle microtubules and activating MAD2, the effector protein of spindle assembly checkpoint (SAC), while MG132 and caffeine activate MPF directly. The increased MPF activity activates RhoA, which then activates MAPK. Although the elevated MAPK activity recruits actin on its own, the increased MPF activity recruits myosin in a RhoA- and MAPK-dependent manner, to induce ooplasmic protrusion. The activated SAC effector proteins prevent cyclin B degradation by binding Cdc20, and hence restraining activation of the Cdc20-Anaphase Promoting Complex (APC) (Musacchio and Salmon, 2007; Visconti et al., 2010). Because the retrieval of demecolcine-induced protrusions have been found involving both the reassembly of the disintegrated spindle and a marked decrease in MPF and MAPK activities after drug withdrawal (Lan et al., 2008), the fact that spindle assembly would take time might help to explain why the demecolcine-induced protrusion lasted longer than the caffeine- or MG132-induced protrusions because the spindle did not disassemble in caffeine or MG132-treated oocytes. However, it was difficult to understand why caffeine induced more stable protrusions than MG132 did. Our hypothesis was as follows. As caffeine increases MPF activity by inhibiting Myt1/Wee1 and dephosphorylating Cdc2 (Kikuchi et al., 2000; Smythe and Newport, 1992), inactivation of the activated MPF following caffeine withdrawal would rely on cyclin B degradation, which takes hours during the normal cell cycle. On the other hand, treatment with MG132, the specific inhibitor of proteasome catalytic activity, would prevent degradation of the ubiquitinated Cdc20 (Ge et al., 2009; Nilsson et al., 2008) as well as cyclin B (Glotzer et al., 1991; Josefsberg et al., 2000). The accumulated Cdc20 would then accelerate cyclin B degradation by activating more Cdc20-APC after oocytes release from MG132 inhibition. However, the present results revealed no difference in persistence between demecolcine- and caffeine-induced protrusions. Taken together, the results suggest the possibility that the duration of chemical-induced ooplasmic protrusions is determined mainly by the time taken for the inactivation of MPF/MAPK rather than by the time for the reassembly of the disassembled spindle.
In this study, although both demecolcine and caffeine induced ooplasmic protrusions successfully in both goat and pig oocytes, the mouse oocyte responded poorly to both drugs. Mouse oocytes also responded poorly to treatment with demecolcine for chemical enucleation, with an optimized enucleation rate of only 21% (Gasparrini et al., 2003). Induction of ooplasmic protrusions in goat oocytes necessitated an increase in both MPF and MAPK activities; although the elevated MAPK activity recruited actin on its own, the increased MPF activity recruited myosin in a RhoA- and MAPK-dependent manner (Wu et al., 2010). The mouse oocyte is different from the goat oocyte in that the actin domain responsible for ooplasmic protrusion is enriched during maturation not after chemical treatment of matured oocytes. Furthermore, although the actin domain disappeared from goat oocytes (Lan et al., 2008), it persisted in mouse oocytes (Liu et al., 2002) after cytochalasin B treatment. Although the MPF activity in mouse oocytes might have increased as caffeine would activate MPF directly and spindle disintegration was observed following demecolcine treatment (data not shown), it is not known whether the activities of MAPK and RhoA increased as well after the treatment and whether the mouse oocyte uses a different mechanism to regulate ooplasmic protrusion. However, Costa-Borges et al. (2009) obtained high rates (84%) of protrusion formation in mouse oocytes after optimized treatments with demecolcine. The difference is that they used KSOM while we used mD-PBS as the basic medium for oocyte treatment. The highest rate of ooplasmic protrusion we obtained with KSOM was only 15% (data not shown), much lower than that obtained with mD-PBS. Strain-specific variations in the rate of oocyte enucleation were detected following chemical enucleation treatment with demecolcine; CF1 oocytes were more efficiently enucleated relative to B6D2F1 oocytes (Ibanez et al., 2003). Contrary to this result, however, Costa-Borges et al. (2009) were unable to observe the mouse strain-specific effect on the efficiency of the anti-mitotic treatments for ooplasmic protrusion.
In summary, we have studied the feasibility to use caffeine or MG132 for chemical-assisted enucleation of goat, pig, and mouse oocytes. Experiments using goat oocytes showed that rates of enucleation, cell fusion, and blastulation were all similar among caffeine-, MG132-, and demecolcine-assisted enucleation but significantly higher than mechanical enucleation. Similar rates of pregnancy and live births were obtained with caffeine- and demecolcine-assisted enucleation. Although the caffeine-induced protrusions persisted more than 2 h, the MG132-induced ones began to disappear at 0.5 h after treatment. Optimal treatments with caffeine efficiently induced ooplasmic protrusions in pig oocytes but mouse oocytes responded poorly to both caffeine and demecolcine. Although it is concluded from the data that caffeine can be used for enucleation of goat and pig oocytes with similar results as demecolcine, the mechanisms for the limited persistence of MG132-induced protrusions and for the refractoriness of mouse oocytes to the protrusion-inducing drugs need further investigations. Such investigations will not only promote the application of the chemical-assisted enucleation technique to more species by selecting more suitable chemicals but also they will contribute to our understanding of the dynamic regulation of the actin cytoskeleton, which plays a crucial role in the control of various cell movements including cell polarization but is facing many problems unsolved due to a lack of suitable in vitro models (Samaj et al., 2004; Villalonga and Ridley, 2004).
Footnotes
Acknowledgments
This study was supported by grants from the China National Project of Transgenics (Nos. 2009ZX08008-006B and 2008ZX08010-001), the China National Natural Science Foundation (Nos. 30771556 and 30972096), and the National Basic Research Project (Nos. 2007CB947403 and 2006CB944003) and the National “863” Project (Nos. 2008AA10Z160) of the China Ministry of Science and Technology.
Author Disclosure Statement
The authors declare that no conflicting financial interests exist.