
Abstract
Abstract
The generation of defined somatic cell types from pluripotent stem cells represents a promising system for many applications for regenerative therapy or developmental studies. Certain key developmental genes have been shown to be able to influence the fate determination of differentiating stem cells suggesting an alternative differentiation strategy to conventional medium-based methods. Here, we present a system allowing controlled, directed differentiation of embryonic stem cells (ESCs) solely by ectopic expression of single genes. We demonstrate that the myogenic master regulator myoD1 is sufficient to induce formation of skeletal muscle. In contrast to previous studies, our data suggest that myoD1-induced differentiation is independent of additional differentiation-inducing or lineage-promoting signals and occurs even under pluripotency-promoting conditions. Moreover, we demonstrate that single gene-induced differentiation enables the controlled formation of two distinct cell types in parallel. By mixing ES cell lines expressing myoD1 or the neural transcription factor ngn2, respectively, we generated a mixed culture of myocytes and neurons. Our findings provide new insights in the role of key developmental genes during cell fate decisions. Furthermore, this study represents an interesting strategy to obtain mixed cultures of different cells from stem cells, suggesting a valuable tool for cellular development and cell–cell interaction studies.
Introduction
Until now, numerous differentiation protocols have been established to generate various cell types from ESCs (Loebel et al., 2003). In general, these strategies consist of a multistep procedure combining the induction of random differentiation with the addition of specific factors, for example, cytokines or growth factors to enhance the formation of a specific lineage. The disadvantage of such multistep procedures is the fact that each component has to be determined in an empirical and thus time-consuming way. Furthermore, each parameter may have different side effects (Schuldiner et al., 2000) rendering the differentiation process susceptible to variability. Additionally, the generation of different cell types requires distinct methods, making it complicated to obtain a mixed culture of different cell types. Such cocultures, however, could provide a useful system to study the interactions between distinct cell types during differentiation.
An alternative approach to direct the fate of stem cells toward a specific lineage is ectopic expression of key developmental genes, so-called master regulators (MRs). It has been shown that lineage-specific MRs can influence the cell fate choice of differentiating ESCs toward a specific cell type (Kanda et al., 2003; Zhao et al., 2004). The advantage of these approaches is the fact that fate determination is partially mediated by an internal signal, namely, the expression of a certain gene, albeit external signals like, for example, embryoid body formation appear to be required to induce and promote differentiation. If, however, external signals could be omitted, the complete differentiation process would depend only on the internal signal of MR expression. Thus, it should be possible to induce directed formation of certain cell types independent from culture conditions.
Based on this idea, we have established a differentiation strategy based on the expression of single MR genes (Thoma et al., 2011; Thoma et al., manuscript in revision). Here, we present a new differentiation system that allows the formation of skeletal muscle from mouse ESCs (mESCs) only by ectopic expression of the myogenic transcription factor myoD1 in the absence of any additional signals. Moreover, we demonstrate that this approach enables the generation of myocytes in parallel to a second MR-induced neuronal differentiation process. It therefore represents a valuable tool to induce directed differentiation of stem cells in a robust and simple way.
Materials and Methods
Plasmids
For generation of the MR induction vectors, a CMV promoter and a SV40 polyadenylation signal were inserted in the pminiTol2/MCS vector (kind gift from S. Ekker) containing the tol2 recognition sites (pMTCpA). The coding sequence of eGFP-Zeo, a fusion protein of eGFP and the zeocin resistance gene, was amplified by PCR with primers containing flanking lox sites, and the PCR product was cloned into pMTCpA resulting in pMTC-eGFP-Zeo. The CMV promoter was exchanged by the ef1a1 promoter (pMTE-eGFP-Zeo). Then, the coding sequence of the MR genes ngn2 and myoD1, respectively, were amplified by PCR and inserted in pMTE-eGFP-Zeo resulting in pMTE-eGFP-Zeo-MR. Puromycin resistance gene and 2A sequence as well as the coding sequence of CreER were amplified by PCR and cloned into pMTE-eGFP-Zeo-MR resulting in pMTE-eGFP-CreER-P2A-MR (subsequently referred to as Cre-myoD1 and Cre-ngn2, respectively).
Cell culture
Mouse 129/Ola-derived IB10 ES cells (a subclone from E14 ES cells; kindly provided by A. Mueller) were grown at 37°C, 5% CO2 on gelatin coated wells in DMEM without phenolred with stable glutamine (2 mM) and sodium pyruvate (1 mM), 10% fully defined fetal bovine serum (FBS) (PAA, Cat. No.: A15-151, Lot No.: A15108-1479), nonessential amino acids (0.1 mM), penicillin/streptomycin, β-mercaptoethanol (0.1 mM), and leukemia inhibitory factor (LIF) (1000 U/mL). The same batch of FBS was used for all experiments. Recombinant murine LIF was produced in Escherichia coli (strain: BL21 DE3 plysS) using a GST-Tag purification system. GST Tag was removed after purification. The same batch of recombinant murine LIF was used for all experiments.
The tol2 transposase system (Kawakami and Noda, 2004) was used for generation of transgenic ES cell lines. Cells were transfected using the Fugene HD Reagent (Roche, Indianapolis, IN) with a vector containing the coding sequence of the tol2 transposase under control of the CMV promoter and the Cre-MR vectors at a ratio of 2:1. After selection with zeocin (100 μg/mL) for 10 days single colonies were picked, expanded, and checked for correct function of the induction constructs. One clone was chosen for further analysis for each cell line.
To induce ectopic expression of MR genes, transgenic cell lines were treated with 4-hydroxytamoxifen (4OHT) at a concentration of 1 μM for 18 h. Control cells were treated with the same volume of 100% ethanol instead of 4OHT. During all differentiation assays, medium was changed every day. Puromycin selection was started at day 2 postrecombination (2dpr) at a concentration of 1 μg/mL and was raised to 2 μg/mL at 3dpr.
For coculture experiments, E14-Cre-ngn2 cells were seeded and E14-Cre-myoD1 cells were added on the following day so that final ratio of cell numbers was about 2:1. After attachment of E14-Cre-myoD1 cells, 4OHT treatment was performed as described above. For cell tracker labeling, 1 day after seeding, E14-Cre-myoD1 cells were incubated in phosphate-buffered saline (PBS) containing 1 μg/mL cell tracker CM-DiI (Molecular Probes, Eugene, OR) for 5 min at 37°C followed by 10 min at 4°C. Cells were washed two times with PBS and fresh medium was added. The following day, E14-Cre-ngn2 cells were seeded so that final ratio of cell numbers was about 2:1 (E14-Cre-myoD1:E14-Cre-ngn2). The next day, 4OHT treatment was performed as described above.
RT-PCRs
Total RNAs were isolated from cell cultures using the peqGold TriFast RNA isolation reagent (Peqlab). To exclude gDNA contamination DNAseI digestion (Fermentas, Hanover, MD) was performed prior to cDNA synthesis (Fermentas). Polymerase chain reaction (PCR) was run from 25 ng cDNA using the following primers: ef1a1 5′- GGTGACAACATGCTGGAGCCAAGTG-3′, 5′-CCCACAGGGACAGTGCCAATGC-3′; endomyoD1 5′-CTACGACACCGCCTACTACAGTGA-3′, 5′- CCTCTGCTGCTGCAGTCGATCT-3′, myf5 5′-ACCTCCAACTGCTCTGACGGCATG-3′, 5′-TGTGTCCTGAAGAGCCAGCTCGGAT-3′; myoG 5′- AGGAGCGCGATCTCCGCTACAGA-3′, 5′-GACATATCCTCCACCGTGATGCTGT-3′; myoT 5′-GTGTGACCACGTGTAACACACGATTAGA-3′, 5′- GGATTGAGCTGCCAGGCGCTGAA-3′; svep1 5′- GTCGGCATTCACATACGGCAGTAAG-3′, 5′- CTGTATCCGTTCTCACAGGAGTACGA-3′; oct4 5′-CACGAGTGGAAAGCAACTCA-3′, 5′-AGATGGTGGTCTGGCTGAAC-3′, nanog 5′-AAGTACCTCAGCCTCCAGCA-3′, 5′- GTGCTGAGCCCTTCTGAATC-3′; afp 5′-CTCAGCGAGGAGAAATGGTC-3′, 5′- GGTGATGCATAGCCTCCTGT-3′, insulin 5′-ATTGTTTCAACATGGCCCTGT-3′, 5′- CTTGTGGGTCCTCCACTTCAC-3′; pax6 5′-GAAGCGGAAGCTGCAAAGAA-3′, 5′- GGAGTGTTGCTGGCCTGTCT-3′; NeuN 5′-AGGACTACTCCGGCCAGACC-3′, 5′- TAGTCGTTTGGGCTGCTGCT-3′.
Positive controls were cDNAs from muscle tissue and mouse embryos (embryonic day 14.5) for myoblast markers, brain for neuronal markers, pancreas for endodermal markers, and embryonic stem cells for pluripotency markers. Each experiment was performed at least three times.
Immunofluorescence staining
Immunofluorescence staining was performed as previously described (Wagner et al., 2009) using anti-Skeletal Myosin antibody (Sigma, St. Louis, MO), anti-Desmin antibody (Sigma), anti-Tuj1 antibody (Novus Biologicals, Littleton, CO), and Hoechst 33258 (Molecular Probes). For microscopy, a Leica DMI6000B inverted microscope was used. All images were analysed using ImageJ software.
Results and Discussion
To establish a differentiation system based solely on the expression of a single gene we chose myoD1 as myogenic MR. MyoD1 is involved in the determination of skeletal myoblasts (Rudnicki et al., 1993) and is able to convert various somatic cell types into skeletal muscle (Davis et al., 1987; Weintraub et al., 1989). Moreover, ectopically expressed myoD1 enhances myogenic differentiation of ESCs, albeit in ESCs the cell fate determination of myoD1 often appears to depend on additional changes of culture conditions, for example, embryoid body formation in low-mitogen containing medium (Dekel et al., 1992). However, as myoD1 is sufficient to convert somatic cells to skeletal muscle in the absence of additional changes in culture conditions (Choi et al., 1990), we reasoned that it should also be sufficient to induce muscle formation in ESCs without additional signals.
To establish a highly controllable and efficient differentiation system we generated a MR induction system with all components located on a single vector (Fig. 1). In this system, 4OHT addition leads to the activation of Cre recombinase (Feil et al., 1996), resulting in the excision of the GFP open reading frame and subsequent translation of the MR and the puromycin resistance gene. Thus, this system allows homogenous induction of MR expression and subsequent selection of MR expressing cells.

Master regulator induction construct. Activation of CreER recombinase by 4OHT treatment results in excision of the open reading frames of CreER and GFPzeo and subsequent translation of the MR and the puromycin resistance gene.
A clonal ES cell line (E14-Cre-myoD1) transgenic for the myoD1 MR induction construct was generated. Cells were treated with 4OHT to induce ectopic expression of myoD1. No additional changes to culture conditions were performed except the withdrawal of LIF from the medium, as this cytokine is known to inhibit myoblast differentiation (Jo et al., 2005; Sun et al., 2007).
At day 1 after 4OHT treatment, successful recombination could be confirmed by loss of GFP signal (Suppl. Fig. 1A and B; see online supplementary data at www.liebertonline.com/cell). Nine days post-recombination (dpr), remaining GFP-positive cells had been eliminated by puromycin selection resulting in a pure culture of myoD1 expressing cells (Suppl. Figure 1C and D).
Eight dpr, cells with skeletal muscle morphology were detected in 4OHT-treated cultures (Suppl. Fig. 1C and Suppl. Fig. 2). These cells stained positive for skeletal myosin and occasionally contained several nuclei (Fig. 2A–D). On the molecular level, activation of the muscle-specific genes myf5 (Ott et al., 1991), myogenin (myoG) (Wright et al., 1989), myotillin (myoT) (Mologni et al., 2001), svep1 (Shefer and Benayahu, 2010), as well as endogenous myoD1 was detected (Fig. 2E) in 4OHT-treated cells, whereas mock-treated cells did not show any detectable expression of these genes. Thus, we conclude that myoD1 expression induced a myogenic differentiation process.
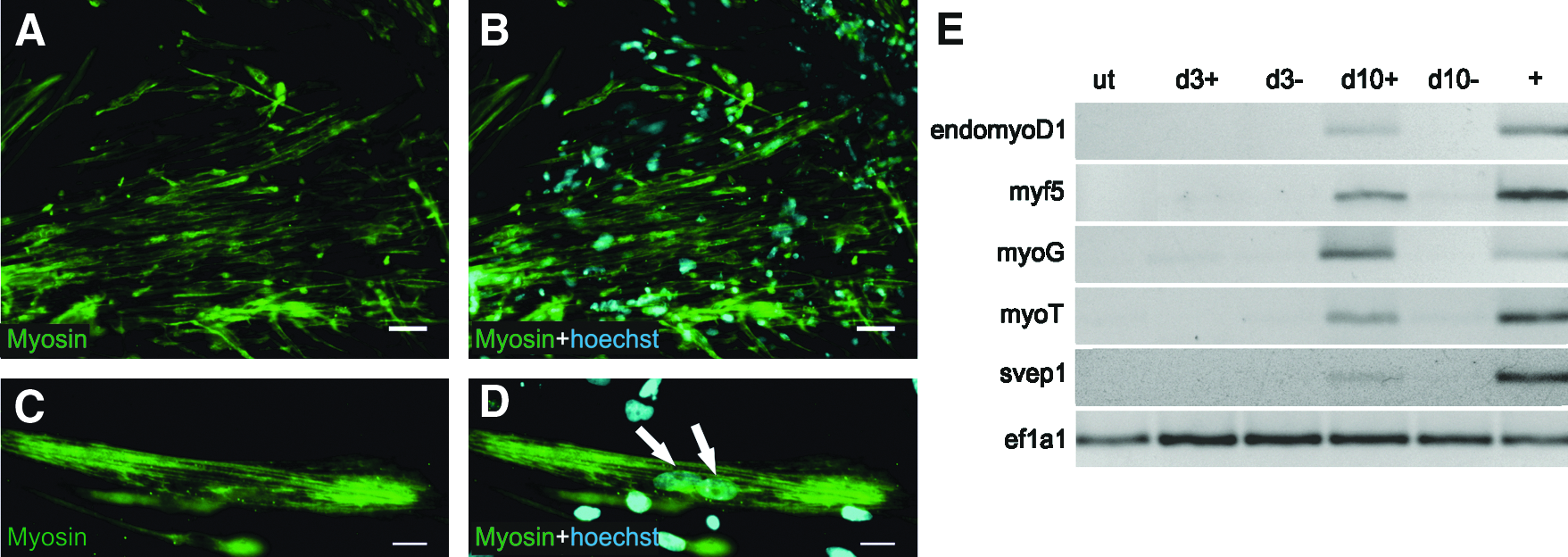
Myogenic differentiation of E14-Cre-myoD1 ES cell line upon ectopic expression of myoD1 in the absence of LIF. (
Next, we wanted to test if myoD1 is also able to induce muscle formation in ESCs under stem cell growth conditions. Therefore, myoD1 expression was induced in the presence of LIF. In LIF containing medium, ectopic expression of myoD1 resulted in the formation of elongated cells expressing the myoblast marker Desmin (Fürst et al., 1989) (Fig. 3A–D). However, no multinucleated cells were detected and cells did not express Myosin (data not shown). RT-PCR revealed upregulation of myogenin, myotillin, and endogenous myoD1 (Fig. 3E). No activation of svep1 and myf5 was observed. These data indicate that even in the presence of LIF, ectopic expression of myoD1 resulted in myogenic differentiation, albeit this process did not proceed as far as under LIF-free conditions. This is in agreement with previous results demonstrating that LIF enhances proliferation and blocks differentiation of myoblasts (Jo et al., 2005; Sun et al., 2007). Nevertheless, our results indicate that myoD1 can act as MR to induce a directed differentiation process in mESCs without the need for any additional differentiation-inducing or lineage-promoting signals. However, the withdrawal of LIF is required for further differentiation of myoblasts.

Myogenic differentiation of E14-Cre-myoD1 ES cell line upon ectopic expression of myoD1 in complete stem cell medium containing LIF. (
Ten dpr, 4OHT-treated cultures did not contain GFP-positive cells (Suppl. Fig. 1C and D) and thus represented an almost pure population of myoD1 expressing cells. As nevertheless nonmyoblastic cells were detectable, we performed RT-PCR for pluripotency (Oct4, Nanog), ectodermal (pax6, NeuN), and endodermal markers (insulin, alpha fetoprotein) to determine the identity of these cells (Suppl. Fig. 3). We did not detect a clear induction of nonmesodermal markers in 4OHT-treated cells compared to mock-treated cells, as it was the case for the myoblast specific markers (Fig. 2E and 3E). Endodermal markers were slightly expressed (insulin, afp), possibly due to random differentiation. Pluripotency markers nanog and oct4 could still be detected in 4OHT and mock-treated cells, indicating the presence of remaining stem cells. Thus, one can assume that myoD1 expressing ESCs either undergo myoblastic differentiation or remain in a stem cell-like state. It will be interesting to analyze what determines a possible differential responsiveness of ESCs to ectopic myoD1 expression.
Until now, our findings suggest that myoD1 is sufficient to induce the formation of skeletal muscle in pluripotent stem cells. As the whole process appears to be driven exclusively in an endogenous and probably in a cell autonomous manner, we assumed that myoD1-induced differentiation should also occur in parallel to another differentiation process. To test this hypothesis, we used an ES cell line (E14-Cre-ngn2) transgenic for the MR induction construct with the coding sequence of myoD1 replaced by that of the neural transcription factor ngn2 (Sommer et al., 1996). Upon induction of ectopic ngn2, E14-Cre-ngn2 cells differentiate into functional neurons under stem cell growth conditions in the presence and in the absence of LIF (Thoma et al., manuscript in revision). To assess whether MR-induced differentiation allows the formation of two different cell types in parallel, E14-Cre-myoD1 cells and E14-Cre-ngn2 cells were seeded in one cell culture dish. Ectopic expression of myoD1 and ngn2, respectively, was induced by 4OHT treatment and cells were cultured in stem cell medium without LIF to facilitate the formation of skeletal muscle cells from E14-Cre-myoD1 cells. Ten dpr, Myosin-positive myoblasts, and Tuj1 positive neurons were detected in close vicinity suggesting that myoD1-induced myogenic differentiation and ngn2-induced neuron formation had occurred in parallel (Fig. 4A–C). To confirm that each cell type arose from the ES cell line expressing the corresponding MR, the experiment was repeated with E14-Cre-myoD1 cells labeled with a fluorescent cell tracker. Numerous cell tracker positive cells stained positive for the myoblast marker Desmin, whereas no cell tracker labeled Tuj1-positive neuron could be detected (Fig. 4D–I). These observations confirm that myoD1 expressing ESCs gave only rise to skeletal muscle cells. Furthermore, RT-PCR analyses of ngn2-expressing cells revealed no activation of muscle-specific genes (Suppl. Fig. 4) excluding the possibility of myoblasts arising from ngn2-expressing ESCs.
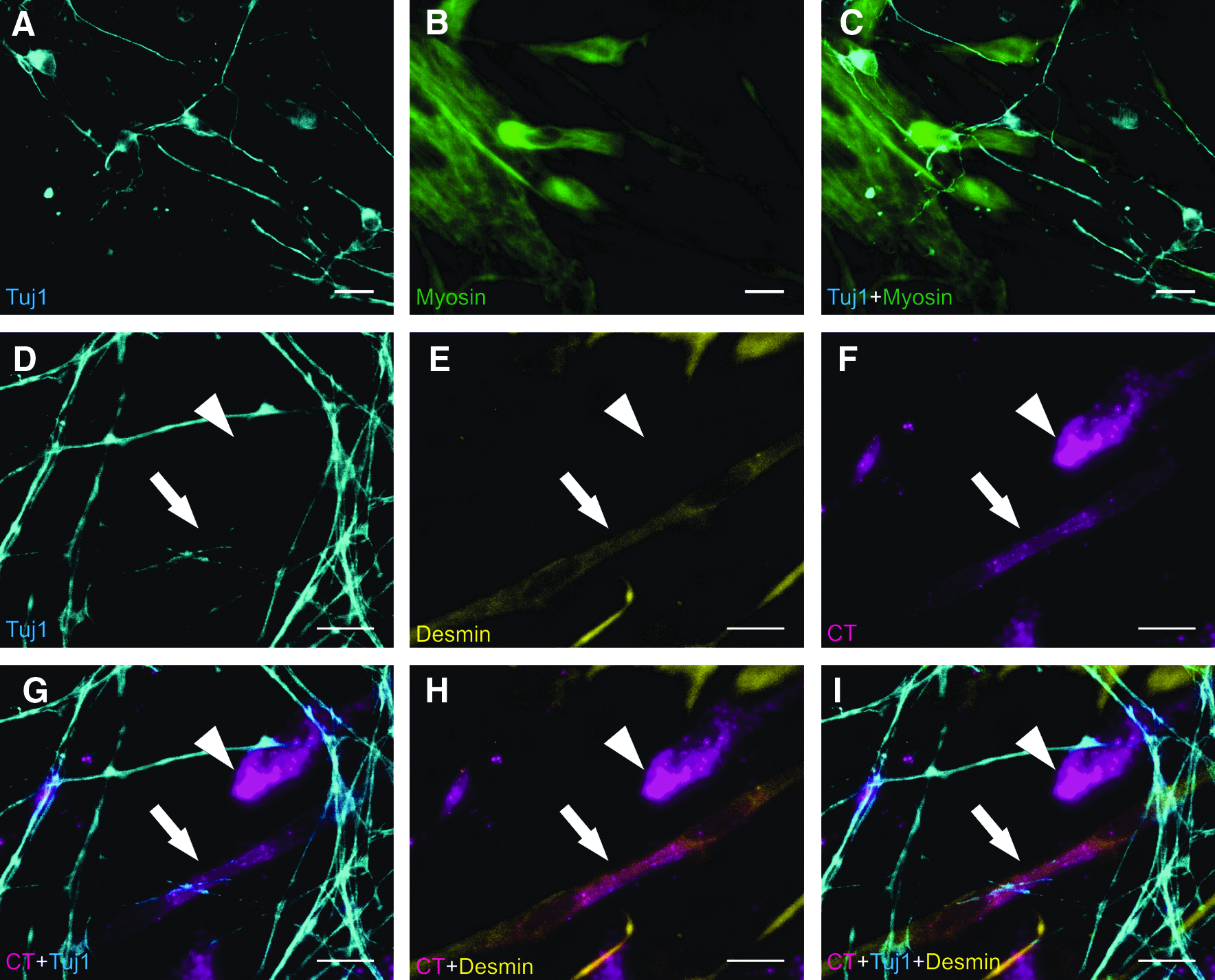
Parallel formation of neurons and skeletal muscle cells using MR-induced differentiation. (
Altogether, these results indicate that MR-induced differentiation allows the formation of two unrelated cell types—skeletal muscle cells and neurons—in parallel by the use of two suitable MR genes: myoD1 and ngn2. Thus, MRs appear to be sufficient to determine the cell fate decision of stem cells largely independent of external signals and processes. These findings highlight the strength of defined key developmental genes in cell fate determination. Furthermore, they suggest MR-induced differentiation as a promising approach to induce directed differentiation in a very robust way and potentially in parallel to other differentiation processes. This strategy could be applied to generate multicellular cultures of distinct cell types as model for interaction and differentiation studies.
Footnotes
Acknowledgments
This work was supported by the Boehringer Ingelheim Fonds for basic medical research. We thank A. Mueller for providing the E14 cell line. We thank F. Guillemot for a construct containing the ngn2 coding sequence. We thank R. Hock for a kind gift of anti-Myosin and anti-Desmin antibodies.
Author Disclosure Statement
The authors declare that no conflicting financial interests exist.
References
Supplementary Material
Please find the following supplemental material available below.
For Open Access articles published under a Creative Commons License, all supplemental material carries the same license as the article it is associated with.
For non-Open Access articles published, all supplemental material carries a non-exclusive license, and permission requests for re-use of supplemental material or any part of supplemental material shall be sent directly to the copyright owner as specified in the copyright notice associated with the article.