
Abstract
Abstract
Somatic cell nuclear transfer (SCNT) is the injection of a donor nucleus into an enucleated egg. Despite the use of this technology for many years in research, it is still quite inefficient. One of the causes for this is thought to be incorrect or incomplete genome reprogramming. Embryos produced by nuclear transfer (cloned embryos) very often present abnormal epigenetic signatures and irregular chromatin reorganization. Of these two issues, the issue of chromatin rearrangements within the nuclei after transfer is the least studied. It is known that cloned embryos often present pericentromeric heterochromatin clumps very similar to the chromocenters structures present in the donor nuclei. Therefore, it is believed that the somatic nuclear configuration of donor nuclei, especially that of the chromocenters, is not completely lost after nuclear transfer, in other words, not well reprogrammed. To further investigate pericentromeric heterochromatin reorganization after nuclear transfer, we decided to study its rearrangements in cumulus-derived clones using several related epigenetic markers such as H3S10P, H3K9me3, and the double marker H3K9me3S10P. We observed that two of these markers, H3S10P and H3K9me3S10P, are the ones found on the part of the pericentromeric heterochromatin that is remodeled correctly, resembling exactly the embryonic heterochromatin configuration of naturally fertilized embryos. Conversely, H3K9me3 and heterochromatin protein 1 beta (HP1β)-associated protein were also detected in the perinuclear clumps of heterochromatin, making obvious the maintenance of the somatic epigenetic signature within these nuclear regions. Our results demonstrate that H3S10P and H3K9me3S10P could be good candidates for evaluating heterochromatin reorganization following nuclear reprogramming.
Introduction
It is thought that these undesirable results are due to inappropriate genome reprogramming in this type of embryo and aberrant epigenetic status, such as abnormal DNA methylation and irregular histone modification patterns (Kang and Roh, 2011). However, most of the reports have focused on the presence/absence of epigenetic modifications in reconstructed embryos and few have analyzed the impact of the inadequate epigenetic status on chromatin reorganization within the resulting nuclei. In previous studies, we demonstrated such chromatin rearrangement abnormalities in cloned embryos after ESC and cumulus cell nuclear transfer using a marker of pericentromeric heterochromatin, heterochromatin protein 1 beta (HP1β) (Maalouf et al., 2009; Martin et al 2006a).
After fertilization, both parental genomes reorganize to form pronuclei, which display a peculiar distribution of pericentromeric heterochromatin in which the centromeres are distributed mostly around the nucleolar precursor bodies (NPBs,) forming a ring-like structure, while the rest of the chromosomes most probably stretch out to the periphery of the nucleus (Martin et al., 2006b). This “cartwheel” organization, which is exclusive to that stage, has been suggested to maintain transcriptional silencing during parental genome maturation.
In embryos obtained by nuclear transfer, it has been shown that pericentromeric heterochromatin is rapidly reorganized as in naturally fertilized embryos (Maalouf et al., 2009; Martin et al., 2006a; Merico et al., 2007). However, reprogramming after nuclear transfer is not perfect, and aberrations are quite frequent: Remains of somatic-like heterochromatin clumps are often observed in late one-cell and early two-cell clones (Maalouf et al., 2009; Martin et al., 2006b). These heterochromatin clumps are very similar to the chromocenters seen in the nucleus of somatic cells. Chromocenters are formed by the clustering of various centromeres of different chromosomes, being basically constituted by pericentromeric and the bordering centromeric heterochromatin (Alcobia et al., 2000; Alcobia et al., 2003).
Interestingly, we have found a link between the developmental inefficiency of cloned embryos and aberrant chromatin reprogramming. We observed that ESC nuclei, which give a higher rate of survival to term after cloning, undergo better remodeling after nuclear transfer than cumulus cell nuclei (Maalouf et al., 2009). Incubation of cloned mouse embryos in the very early hours after transfer with a histone deacetylase (HDAC) inhibitor improved the structural remodeling of pericentric heterochromatin at the one-cell stage and dramatically increased the rate of full term development (a 10-fold increase) (Maalouf et al., 2009).
Altogether, these results indicated that the nuclear configuration of donor nuclei, and especially pericentromeric heterochromatin clustering into chromocenters, was not completely lost after nuclear transfer and that it impaired development. To further investigate this hypothesis, we decided to follow pericentromeric heterochromatin rearrangements in cumulus-derived clones using various epigenetic markers. Therefore, we performed immunostainings on the two most obvious and important stages after meiosis resumption, i.e., the one-cell stage with formation of the pronuclei and the two-cell stage when embryonic genome activation normally occurs in the mouse.
First, we analyzed phosphorylation of histone H3 at serine 10 (H3S10P). In mammalian cells, phosphorylation of histone H3 at serine 10 is evident first in pericentromeric heterochromatin in late G2-interphase cells, spreading throughout the chromosomes arms during prophase, and only being dephosphorylated around late anaphase (Perez-Cadahia et al., 2009). The fact that H3S10P is detected only at the end of interphase and observed in the entire chromosomes lengths led to the conclusion that this epigenetic modification could be related to chromosome condensation (Garcia et al., 2005). However, this posttranslational modification has also been observed outside mitosis, and it is believed that it is involved with gene activation (Drobic et al., 2010; Lim et al., 2004). Some research groups have investigated this epigenetic modification during early mouse embryogenesis. Their reports suggest that H3S10P is linked to pericentromeric heterochromatin (Huang et al., 2007; Teperek-Tkacz et al., 2010; Wang et al., 2006). Recently, we have shown by immunofluorescence in situ hybridization (immuno-FISH) using specific pericentromeric heterochromatin probes that H3S10P indeed marks constitutive heterochromatin during interphase until the four-cell stage in mouse embryos (Ribeiro-Mason et al., submitted).
The chromocenters are also characterized by trimethylation of histone H3 at lysine 9 (H3K9me3), which provides a binding site for HP1β; this complex then induces transcriptional repression and heterochromatinization in the pericentromeric heterochromatin domains (Lachner et al., 2001). In fertilized embryos, these two markers are distributed asymmetrically between maternal and paternal pronucleus; at the pronuclear stage, only the maternal pronucleus contains H3K9me3 and HP1β accumulations (Cowell et al., 2002; Santos et al., 2005). This was confirmed by immuno-FISH experiments showing that both H3K9me3 and HP1β colocalize with pericentromeric heterochromatin probes, especially around the NPBs of the maternal pronuclei (Probst et al., 2007).
Likewise, H3K9me3, HP1β, and H3S10P, the double modification H3K9me3S10P (trimethylation of histone H3 at lysine 9 phosphorylated at serine 10), are also detected in the pericentromeric heterochromatin domains in somatic cells. It was first stated that both modifications coexist on the same histone tail, especially during mitosis, based on the results obtained by in vitro assays and analysis of the in vivo modification pattern of H3 isolated from HeLa cells (Fischle et al., 2003). However, H3K9me3S10P correlation with pericentromeric heterochromatin was later described in somatic cells by peptide competition assays and immunofluorescence experiments (Hirota et al., 2005). In fact, H3K9me3S10P can be detected at late G2 in interphase; it was found to be enriched in the centric and pericentric domains, with a more spotted appearance on the chromosome arms during mitosis (Fischle et al., 2005; Monier et al., 2007). Only one study has mentioned this epigenetic modification in fertilized mouse embryos, showing that upon the first mitosis it was preferentially associated with maternal chromosomes (Hayashi-Takanaka et al., 2009).
In this study, we investigated the status of three epigenetic markers, related to pericentromeric heterochromatin (H3K9me3, H3S10P, and H3K9me3S10P), in cloned mouse embryos at the one- and two-cell stages to find out if one of these markers could be used as another tool to monitor chromatin reorganization after nuclear transfer.
Materials and Methods
Animal care and handling were carried out according to European regulations on animal welfare.
Oocytes and embryos production
C57/CBA F1 female mice, 6–8 weeks of age, were superovulated with 5 IU of pregnant mare serum gonadotropin (PMSG) followed by injection with 5 IU of human chorionic gonadotropin (hCG) 48 h later. For in vivo embryo production, females were placed together with males (one by one) after hCG administration. Embryos were collected in M2 medium containing 1 mg/mL hyaluronidase and then cultured in M16 at 37°C in a humidified atmosphere containing 5% CO2 until fixation for immunofluorescent staining. Fertilization occurred at about 12 h after hCG injection, and this was used as the reference point for embryonic development (hours post-hCG, i.e., hphCG).
Cumulus cell nuclear transfer
Oocytes were prepared by superovulating C57/CBA mice. Superovulation was induced by injecting PMSG (Intervet, 5 IU) and hCG (Intervet, 5 IU) at intervals of 48 h. Oocytes were collected from oviducts 14 hphCG and washed in M2 medium containing 1 mg/mL hyaluronidase. Subsequently, they were incubated in M2 containing 5 μg/mL cytochalasin B and placed in a chamber on the stage of an inverted microscope (Nikon) equipped with micromanipulators (Nikon-Narishige MO-188). The chromatin spindle (visualized under differential interference contrast) was aspirated into the pipette as previously described (Zhou et al., 2000). For nuclear transfer, donor chromosomes were derived from cumulus cells that previously surrounded the oocytes. They were gently aspirated in and out of the injection pipette (inner diameter 7–8 μm) and then microinjected into the cytoplasm of the enucleated oocytes. The nuclear transfer embryos were activated by incubation for 6 h in Ca2+-free medium containing 10 mM Sr2+ and 5 μg/mL cytochalasin B. Embryos with visible nuclei were then considered as activated, transferred into fresh M16 medium, and cultured at 37°C in a humidified atmosphere containing 5% CO2. Embryos were fixed during the first cell cycle [at 4, 5, 8, and 10 h postactivation (hpa)], and early and late two-cell stages (21 hpa and 33 hpa, respectively).
Immunofluorescent staining
The following antibodies were purchased from the indicated companies: rabbit polyclonal antibody against H3S10P (Abcam #5176); mouse monoclonal antibody against HP1β (Euromoedex #MOD-1A9-AS); rabbit polyclonal antibody against H3K9me3 (Upstate #07-523); rabbit polyclonal antibody against H3K9me3S10P (Abcam #5819); fluorescein isothiocyanate (FITC)-conjugated secondary antibody and Cy5-conjugated secondary antibody from donkey (Immunoresearch, Jackson Laboratories).
Embryos in different developmental stages were fixed with 4% paraformaldehyde (PFA) in phosphate-buffered saline (PBS) at 4°C overnight and permeabilized with 0.5% Triton X-100 [15 min, at room temperature (RT)]. The fixed embryos were blocked in PBS containing 2% bovine serum albumin (BSA) (1 h at RT) and incubated overnight at 4°C with the specific first antibody diluted in 2% PBS-BSA (at 1:300 for H3S10P; 1:400 for HP1β; 1:400 for H3K9me3; 1:300 for H3K9me3S10P). The embryos were then washed twice in PBS to remove any first antibody excess. After this step, the embryos were incubated with FITC or Cy5-labeled secondary antibody for 1 h at RT (dilution 1:200). DNA counterstaining was performed with ethidium homodimer 2 or propidium iodide (Invitrogen). Embryos were then postfixed with 2% PFA for 15 min at RT, washed, and mounted on slides with an antifading agent (Citifluor) under coverslips.
High-resolution microscopy
Three-dimensionally (3D)-preserved embryos were observed with either a Carl Zeiss AxioObserver Zl fluorescence microscope equipped with the ApoTome slider or a Zeiss LSM 510 confocal laser scanning microscope (MIMA2 Platform, INRA). On the Apotome, embryos were observed using a 63×Plan-Neofluar oil objective [numerical aperture (NA) 1.3] and single-wavelength light-emitting diodes (LEDs) at 470 nm, 530 nm, and 625 nm (Colibri illumination). Digital optical sections were collected using a Z-series acquisition feature every 1 μm. For the confocal system, embryos were visualized with an oil-immersion objective (Plan Apochromatic 63×NA 1.4), and imaging was performed with lasers at 488-, 535-, and 633-nm wavelengths. Entire embryos were scanned with a distance of 0.37 μm between light optical sections. For intensity profile measurements, line scans were obtained with ImageJ software (http://rsbweb.nih.gov/ij/).
Results
H3S10P distribution pattern in fertilized and cumulus cloned embryos
As previously described (Huang et al., 2007), histone H3 was phosphorylated at serine 10 shortly after fertilization; the decondensing sperm head was already labeled for H3S10P as well as the maternal chromosomes (n=27 at 18 hphCG; Fig. 1). Upon formation of the pronuclei, a diffuse nucleoplasm H3S10P labeling was present in all embryos with some accumulations appearing at the periphery of the NPBs (n=35 early one-cell at 18 hphCG; Fig. 1, arrows). This perinucleolar staining then formed full heterochromatin rings around the NPBs (n=29 at 20 hphCG) that became more intense in late one-cell embryos (n=33 at 26 hphCG; Fig. 1). When entering mitosis of the first cell cycle, H3S10P staining had spread out throughout the whole mitotic chromosomes as on the metaphase II chromosomes (n=25 at 28–30 hphCG; Fig. 1 and data not shown). For early two-cell-stage embryos, the staining was again present uniformly in the nucleoplasm and more intensely on the heterochromatin rings surrounding the NPBs (n=18 at 36 hphCG; Fig. 1). As the second cell cycle progressed, the pericentromeric regions of different chromosomes assembled together to form unique structures called chromocenters, which were strongly labeled with H3S10P (n=48 at 48 hphCG; Fig. 1, arrows). For the second cell cycle, phosphorylation of histone H3 at serine 10 was also detected on the whole chromosomes following the same pattern as seen during the first mitosis (data not shown).
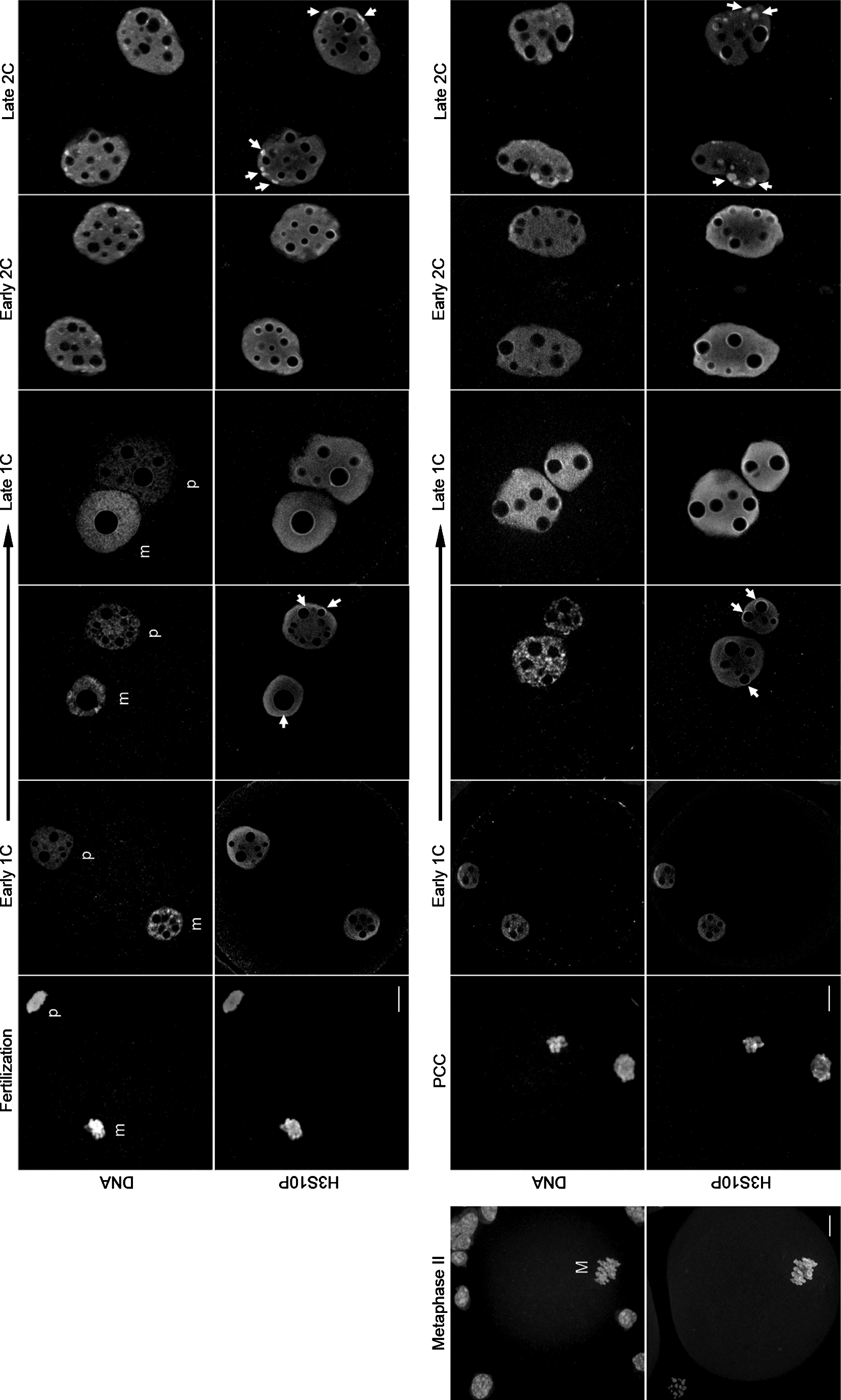
H3S10P detection in fertilized and cloned embryos. Representative images (Apotome single z-sections) of embryos produced by natural fertilization (upper panel) or nuclear transfer (lower panel) stained for H3S10P and DNA, as well as one metaphase II embryo with cumulus cells (M, metaphase/confocal single z-section). The signal seen on the parental genomes (m, maternal; p, paternal) at the beginning of development in fertilized embryos is also accurately observed on the PCC in cloned embryos. During pronuclei formation, both types of embryos present the same diffuse nucleoplasm H3S10P labeling. Signals for H3S10P in the heterochromatin rings around the NPBs then start appearing (arrows). Finally, in late one-cell (1C) stage, the heterochromatin rings are completely formed and a strong staining for H3S10P is seen around the NPBs for both types of embryos. At the two-cell (2C) stage, H3S10P was first detected in the heterochromatin rings around the NPBs (Early 2C) and then on the newly formed chromocenters (Late 2C, arrows). We again observed a similar distribution pattern in fertilized and cloned embryos. Scale bars, 10 μm. PCC, premature chromosomal condensation.
We also performed immunodetection of H3S10P in in vitro–fertilized embryos to identify any abnormality that could have been caused by the embryo culture conditions. On the basis of our observations from fertilization to late two-cell stage, the same distribution pattern for H3S10P was observed in in vivo–fertilized and in vitro–fertilized embryos (data not shown).
We then checked constitutive heterochromatin rearrangements by nuclear reprogramming in cloned mouse embryos produced by somatic cell nuclear transfer (SCNT) with cumulus cells (Fig. 1, lower panel). In these embryos, phosphorylation of histone H3 at serine 10 was detected as early as 2 hpa on the chromatin of the mouse cumulus cell that was undergoing premature chromosome condensation (PCC) inside the enucleated mouse oocytes (n=22; Fig. 1). At 4 hpa, when pseudo-pronuclei are formed, H3S10P recapitulated exactly the same pattern as observed in early fertilized embryos with uniform staining in the nucleoplasm (n=19, early one-cell; Fig. 1). Only three of these embryos (≈16%) showed a slight labeling of the heterochromatin rings surrounding the NPBs. One hour later, 100% of the cloned embryos analyzed had these perinucleolar rings labeled with H3S10P (n=17, 5 hpa; Fig. 1, arrows). At the end of the one-cell stage, all of the cloned embryos showed a strong H3S10P labeling corresponding to that observed in fertilized embryos (n=16 at 7 hpa, n=17 at 8 hpa, and n=29 at 10 hpa; Fig. 1).
For the two-cell stages, all of the cloned embryos had the same H3S10P pattern as seen in fertilized ones (Fig. 1). At 21 hpa (corresponding to early two-cell), H3S10P was observed uniformly in the nucleoplasm, and a strong signal was still seen on the heterochromatin rings at the periphery of NPBs. This staining was then replaced by labeling in the chromocenters forming at late two-cell (33 hpa; Fig. 1, arrows).
Remarkably, cumulus cells had a typical somatic cell pattern for H3S10P and only very few showed a positive signal, i.e., in late G2 and mitosis (Fig. 1). However, 100% of the cloned embryos we observed were positive for this marker over the two first embryonic cycles, suggesting that reprogramming had occurred.
H3S10P and HP1β label distinct types of heterochromatin in early cloned embryos
To evaluate whether H3S10P reprogramming after cloning was concomitant with nuclear remodeling, and especially heterochromatin rearrangements, we then analyzed the colocalization of H3S10P and HP1β. As mentioned before, HP1β has been extensively used as a marker to check pericentromeric heterochromatin distribution, and it was demonstrated that some portion of pericentromeric heterochromatin was accumulating at the nuclear periphery in embryos after nuclear transfer (Maalouf et al., 2009; Martin et al., 2006b; Merico et al, 2007).
We observed a high number of HP1β perinuclear accumulations in the early one-cell stage at 4 hpa, as well as a uniform euchromatic staining, but none of these HP1β foci were labeled with H3S10P (n=11; Fig. 2, arrow). Remarkably, HP1β and H3S10P showed colocalization only when heterochromatin rings appeared around the NPBs. At the late one-cell stage (10 hpa), HP1β was still observed in numerous perinuclear foci but also around all the NPBs. However, H3S10P did not show any colocalization with HP1β in the clumps of heterochromatin found at the nuclear periphery, but only in the ones surrounding the NPBs (n=21; Fig. 2, line scans).

Double immunostaining with H3S10P and HP1β in early cloned embryos. Cloned embryos were stained for H3S10P and HP1β protein at various time points after activation and observed on the Apotome microscope (only single z-sections are shown here). At the one-cell (1C) stage, it appears that HP1β shows accumulation in the nuclear periphery (arrows) whereas a stronger signal for H3S10P is visualized around the NPBs, both at 4 hpa and 10 hpa. At early two-cell (2C) stage (21 hpa), H3S10P and HP1β only show colocalization on the rings of heterochromatin; again the HP1β foci lack a signal for H3S10P. Nearly full colocalization of both markers is seen by the end of the two-cell stage (33 hpa, arrows). Scale bar, 10 μm. Line scans on the right side of the figure show the local intensity distribution of H3S10P and HP1β labeling. In the merged images, the positions and directions of the line scans are indicated by long arrows. Asterisks on the line scans point to several positions that correspond to HP1β foci without H3S10P signal. Hash symbols underline the chromocenters labeled with both HP1β and H3S10P.
At the two-cell stage, HP1β followed the same distribution pattern as previously described. During the early two-cell stage (21 hpa), HP1β was detected in the nucleoplasm, around the NPBs, and in isolated foci. As in one-cell cloned embryos, H3S10P showed almost no colocalization with HP1β in isolated foci but only on NPBs periphery (n=15; Fig, 2, line scans). This difference disappeared by the late two-cell stage (36 hpa) when both markers colocalized mostly within the newly formed chromocenters (n=10; Fig. 2, arrows).
On the basis of these findings, we suspected that cloned mouse embryos have two types of heterochromatin after nuclear transfer: one that is being remodeled, showing H3S10P and HP1β around the NPBs, and another one, with only HP1β at the nuclear periphery. To confirm whether this perinuclear heterochromatin indeed corresponds to nonreprogrammed somatic heterochromatin, we then focused on another typical marker of pericentromeric heterochromatin—H3K9me3.
Comparison with H3K9me3
As expected, only the maternal genome stained for this marker in fertilized embryos, whereas the paternal one showed no labeling (n=18 at 18 hphCG; Fig. 3). This staining was then distributed all over the nucleoplasm and formed partial rings around NPBs (n=21 at 20/21 phCG). At 26 hphCG, we observed full staining of the periphery of the NPBs, and, remarkably, we regularly noticed one clump of H3K9me3 at the nuclear periphery similar to the clumps previously observed with HP1β (n=32; Martin et al., 2006b). We later confirmed that H3K9me3 and HP1β are colocalized within these perinuclear clumps (β=33; data not shown). Finally, we observed that the paternal genome became stained faintly upon entry in mitosis (n=33; Fig. 3), whereas H3K9me3 strongly labeled almost the whole maternal genome, as expected (Puschendorf et al., 2008).
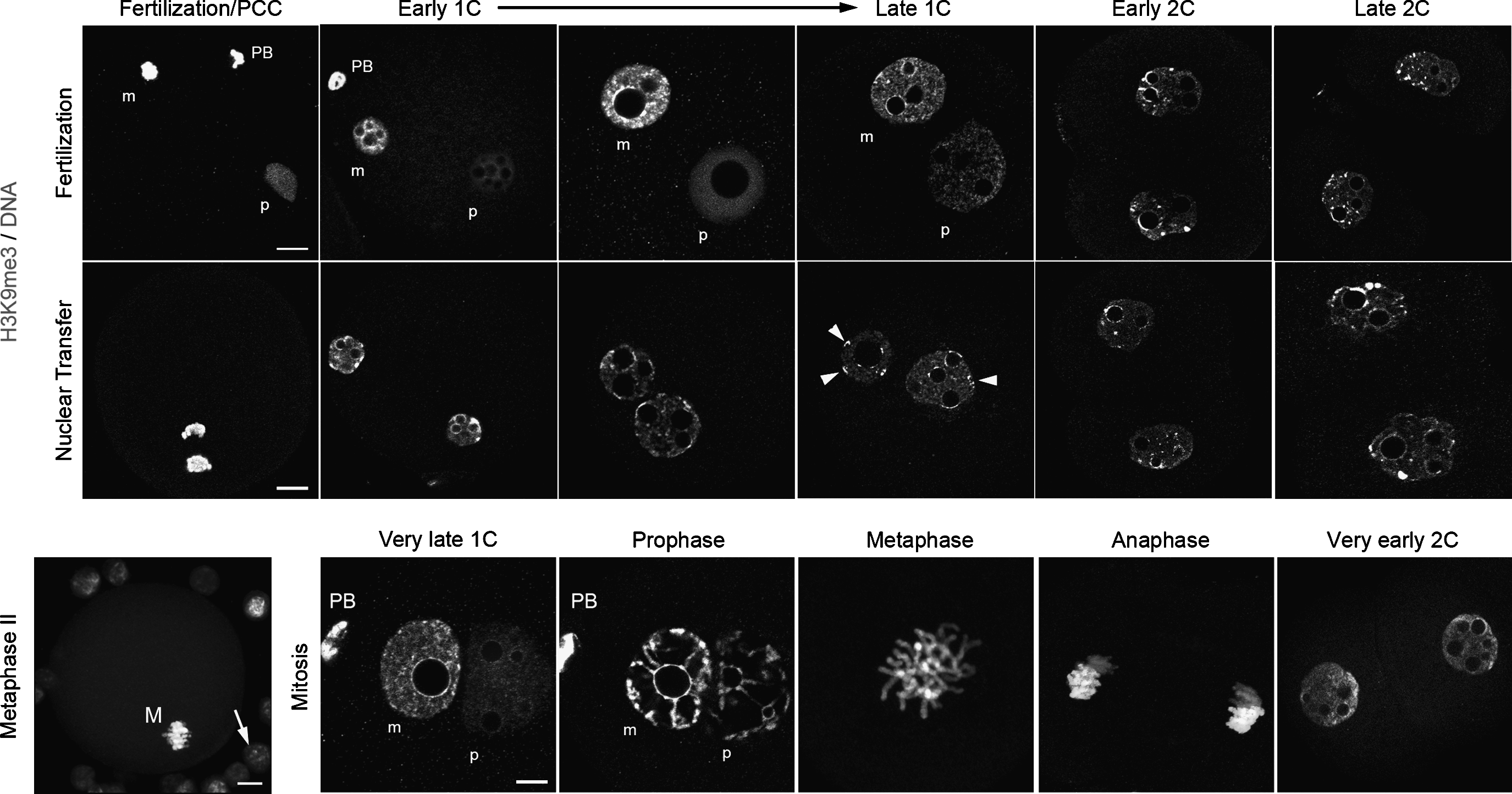
Comparison of H3K9me3 distribution in fertilized and cloned embryos. Naturally fertilized and cloned embryos were stained for H3K9me3 and counterstained for DNA. Representative images from early one-cell (1C) stage until late two-cell (2C) stage shown here are single-sections from z-stacks taken on the confocal microscope. Scale bars, 10 μm. In fertilized one-cell embryos, the two parental pronuclei can clearly be distinguished (m, maternal; p, paternal). To make comparisons easier, we rotated the images, when required, to show the paternal one on the right hand side (PB, polar body). As expected, we observed that H3K9me3 showed parental asymmetry over the whole first-cell stage and even within the two-cell stage nuclei. Upon mitosis, from the very late one-cell stage until the formation of two-cell stage nuclei (≈1 h postcleavage), this parental asymmetry can still clearly be distinguished (note that late one-cell and two-cell are single z-sections, whereas prophase/metaphase/anaphase correspond to z-stack projections). In contrast to fertilized embryos, clones often present perinuclear blocks of H3K9me3-labeled heterochromatin at one-cell (arrowheads), as in the donor cumulus cells that can be seen on the metaphase II oocyte (arrow, M, metaphase).
In comparison, both pseudo-pronuclei formed in early cloned mouse embryos showed H3K9me3 staining and no asymmetry (Fig. 3). At 4 hpa and 6 hpa (n=16 and n=14, respectively), we observed heterogeneous nucleoplasmic staining and partial perinucleolar rings (as in fertilized embryos) but also a high number of H3K9me3 perinuclear foci similar to the ones observed in the cumulus donor cells (Fig. 3). At 10 hpa, H3K9me3 was still present in numerous perinuclear foci, but was then clearly accumulated all around the NPBs (n=25, Fig. 3). Again, this suggested that cumulus inherited heterochromatin was not fully remodeled after nuclear transfer.
As previously described, H3K9me3 followed the same distribution pattern as HP1β in two-cell-stage fertilized and cloned embryos (Figs. 2 and 3; Maalouf et al., 2009; Merico et al., 2007). Asymmetric diffuse staining in the nucleoplasm with few perinuclear accumulations and strong perinucleolar staining was observed at 36 hphCG (n=16 early two-cell; Fig. 3), whereas the nucleoplasm was homogeneously stained at 48 hphCG, with intense labeling appearing on the chromocenter-like structures (n=18, late two-cell). Cloned embryos demonstrated a similar dynamic, with the exception of supplementary isolated foci in early two-cell embryos (n=9 at 21 hpa and n=16 at 36 hpa; Fig. 3). Altogether, it clearly appeared that H3K9me3 staining was very similar to that observed for HP1β, both in one-cell and two-cell cloned embryos, and that it differed partially from H3S10P after nuclear transfer.
H3K9me3S10P as a new marker of nuclear reprogramming
It is well documented that H3K9me3 is a marker of heterochromatin. Conversely, some studies have stated that the double modification H3K9me3S10P could also be a marker of heterochromatin staining chromocenters in G2 phase of somatic cells. Because this double modification had never been carefully investigated in mouse embryos, we analyzed its redistribution after fertilization and nuclear transfer.
In fertilized embryos, H3K9me3S10P could only be detected in the maternal genome upon fertilization (n=14 at 18 hphCG; Fig. 4). Upon formation of the pronuclei, heterochromatin accumulations then appeared around the NPBs (n=14 and n=16 at 20/21 hphCG; Fig. 4). We noticed that H3K9me3S10P intensity decreased from 20 hphCG onward (n=14 at 27 hphCG; Fig. 4). However, in late one-cell embryos, nice heterochromatin rings could be seen in all the female pronuclei (n=15 at 29 hphCG and n=14 at 30 hpCG; Fig. 4). Remarkably, both parental genomes were labeled during mitosis; also the paternal remained more weakly stained and could still be distinguished (n=22 at 29–30 hphCG; Fig. 4, arrows ).
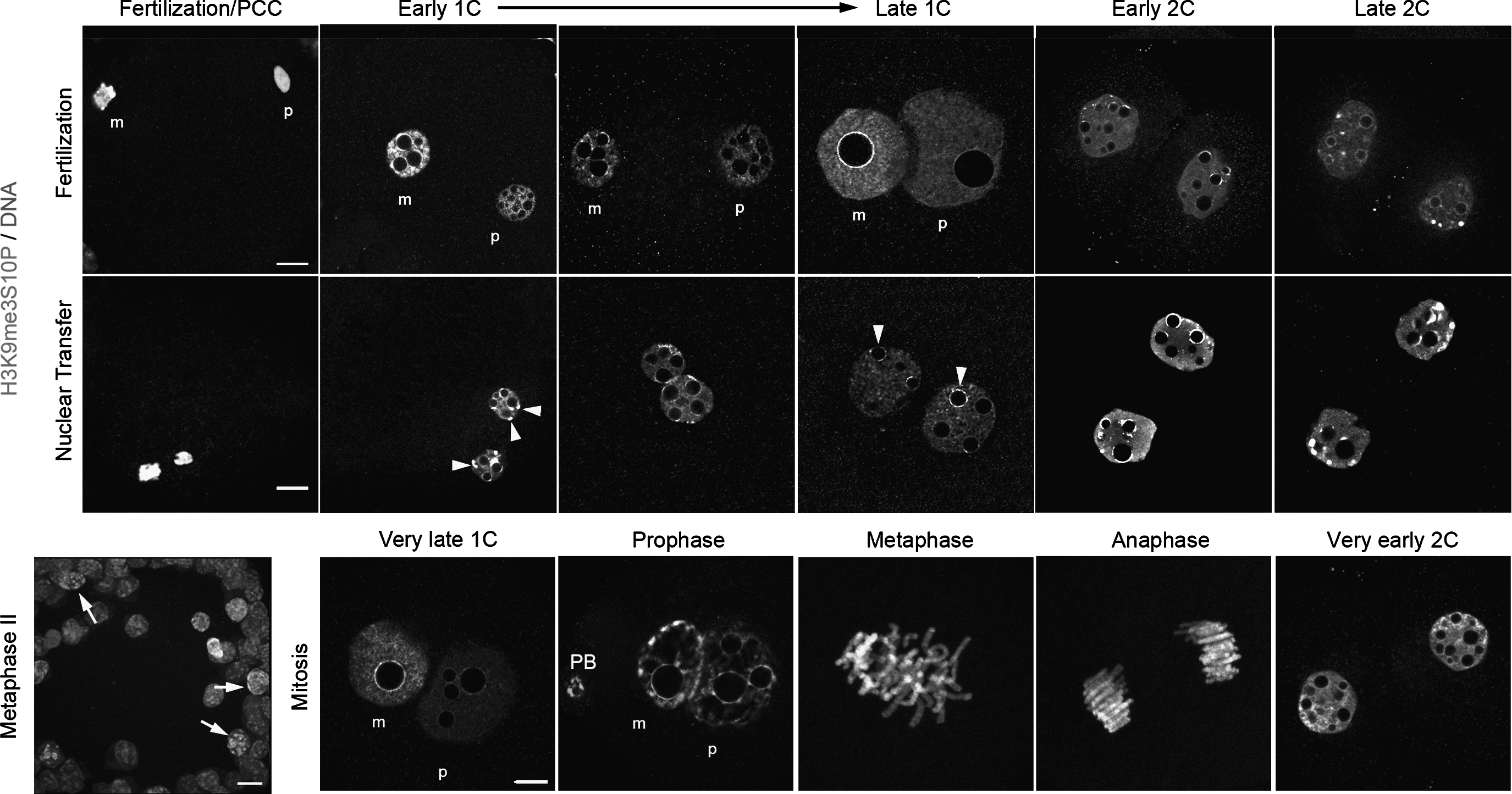
Comparison of H3K9me3S10P distribution in fertilized and cloned embryos. Naturally fertilized and cloned embryos were stained for H3K9me3S10P and counterstained for DNA. Representative images from early one-cell (1C) stage until late two-cell (2C) stage shown here are single-sections from z-stacks taken on the confocal microscope. Scale bar, 10 μm. In one-cell fertilized embryos, the two parental pronuclei can clearly be distinguished (m, maternal; p, paternal). To make comparisons easier, we rotated the images, when required, to show the paternal one on the right hand side (PB, polar body). We observed that H3K9me3S10P epigenetic modification showed maternal specific labeling over the whole first-cell stage. By the late one-cell stage, H3K9me3S10P intensity decreases and the staining remains only on the NPBs periphery (arrowheads). Remarkably, both parental genomes were labeled during mitosis, from the very late one-cell stage until the formation of two-cell stage nuclei ≈1 h postcleavage (note that late one-cell and two-cell are single z-sections, whereas prophase/metaphase/anaphase correspond to z-stack projections). In clones, we observed strong perinuclear blocks of heterochromatin at the beginning of the one-cell stage (arrowheads) but not in late one-cell embryos. These blocks of heterochromatin could also be observed in some donor cumulus cells that can be seen on the metaphase II oocyte (arrows).
Before nuclear transfer, cumulus cells had a similar somatic cell pattern as for H3S10P; only very few showed a positive signal, i.e., in late G2 and mitosis (Fig. 4). However, H3K9me3S10P was present within all the cloned embryos undergoing PCC (n=12 at 2 hpa; Fig. 4), and both pseudopronuclei showed strong staining of heterochromatin clumps just after (n=20 at 4 hpa; Fig. 4, early one-cell, arrowheads). We then observed a clear remodeling of these regions during the first cell cycle (n=20 at 7 hpa; Fig. 4); also the overall intensity seemed to decrease as in fertilized embryos. Finally, H3K9me3S10P was concentrated only on the NPBs periphery in late one-cell cloned (n=20 at 10 hpa; Fig. 4, late one-cell/arrowheads) and not on any perinuclear heterochromatin accumulations, as already observed for H3S10P.
In early and late two-cell stage embryos, the H3K9me3S10P pattern was very similar in fertilized and cloned embryos. Just after cleavage, H3K9me3S10P accumulations were observed mostly around the NPBs and in some isolated foci (n=21 early two-cell at 36 hphCG and n=20 late two-cell at 21 hpa; Fig. 4). This staining was then replaced by clumps on chromocenters in late two-cell embryos (n=14 at 48 hphCG and n=15 at 33 hpa; Fig. 4). The only difference between the two groups was that H3K9me3S10P staining accumulated in one pole of the nuclei (most probably the maternal inherited one), whereas this asymmetry was completely lost in clones.
In conclusion, H3K9me3S10P staining did not correspond to the combination of H3K9me3 and H3S10P staining, demonstrating that these two epigenetic modifications are not always adjacent within the same histone H3 tail in mouse early embryos.
Discussion
One of the main issues with the nuclear transfer technique is genome reorganization of the somatic donor nucleus, a process triggered by nuclear reprogramming, which is thought to be controlled mainly by the enucleated oocyte. For that reason, in this study we have investigated the dynamics of genome restructuring in early mouse embryos derived from cumulus nuclear transfer with an emphasis on pericentromeric heterochromatin-related markers such as H3S10P, H3K9m3, HP1β, and the double modification H3K9m3S10P.
Our results show that the cumulus donor nucleus is remodeled to a certain extent in cloned mouse embryos, as evidenced by the chromatin configuration seen in early one-cell stages as early as 4 hpa (≈18 hp hCG). At this time point, H3S10P showed the same distribution pattern as observed in fertilized embryos. The pseudopronuclei showed uniform staining for H3S10P in the nucleoplasm and strong labeling in the heterochromatin rings around the NPBs from 5 hpa to 10 hpa. However, at 10 hpa (late one-cell) we observed that both H3K9me3 and HP1β accumulated around the NPBs and also in the nuclear periphery, an aberration known to correlate with poor development (Maalouf et al., 2009; Martin et al., 2006b). Differently from these two markers, H3S10P colocalized only with the heterochromatin located around the NPBs. Altogether, this indicates that H3S10P, as opposed to H3K9me3 and HP1β, labels only remodeled pericentromeric heterochromatin located around the NPBs, resembling exactly the normal embryonic heterochromatin arrangement. This also proves that heterochromatin clumps located at the nuclear periphery are unremodeled, maintaining the epigenetic signature of the cumulus cells (with H3K9me3 and HP1β staining on chromocenters).
As for the other epigenetic modification studied, H3K9me3S10P, strong staining was also detected in heterochromatin accumulations in the nuclear periphery, but only at very early stages (4 hpa). Later on (7 hpa/10 hpa), the spatial distribution of H3K9me3S10P shifted and the double modification only colocalized with the heterochromatin rings around the NPBs. We can hypothesize that this double modification was being progressively reprogrammed during the first cell cycle and finally overlapped with the portion of remodeled heterochromatin. In light of these results, we hypothesize that both epigenetic modifications H3S10P and H3K9me3S10P could be used to follow up reprogrammed heterochromatin during nuclear remodeling.
The perinuclear accumulations seen in one-cell stage cloned embryos with H3K9me3 and HP1β staining are probably due to irregular chromatin rearrangement. The reason for this preferential positioning is unknown; however, we can infer that oocytes proteins responsible for nuclear and chromatin organization are involved. It is known that the somatic chromatin configuration must be reshuffled by the reprogramming factors present in the cytoplasm of the enucleated oocyte. Among them are the nuclear lamin filaments that lie on the interface of the nuclear envelope and chromatin that play a major role in nucleoskeleton support, chromatin remodeling, as well as protein recruitment to the inner nucleolus (Hall et al., 2005). It has also been suggested that additional “motor proteins” are in place to assist with chromatin organization, such as nuclear actin in mouse embryos (Nguyen et al., 1998). It is not known how and what makes chromatin move inside the nucleus and if this process happens in a coordinated or random way. However, it might be that lamins and motor proteins are disrupted during the nuclear transfer procedure, and chromatin is misplaced as a result. It would be of great relevance to investigate this hypothesis further to better understand the role of these proteins in nuclear and chromatin organization within the early stages of reprogramming.
In fertilized embryos, it unmistakably appears that H3S10P is a better marker of pericentromeric heterochromatin as compared to other epigenetic markers such as H3K9me3 and H3K9me3S10P, because it is always correlated to this type of heterochromatin in both inherited parental genomes from the very beginning of development. Indeed, both H3K9me3 and H3K9me3S10P epigenetic modifications showed parental asymmetry over the whole first-cell stage. Similarly to H3K9me3 (Cowell et al., 2002; Santos et al., 2005; this study), only the maternal pronucleus is labeled for H3K9me3S10P epigenetic modification in the rings around the NPBs, and a diffuse staining is observed in the nucleoplasm after fertilization. However, a slight decrease in H3K9meS10P intensity is seen starting from 20 hphCG. This decrease most probably corresponds to chromatin decondensation and incorporation of new histones H3 upon the first replication phase, as already described for H3K9me3 (Liu et al., 2004; Wang et al., 2007).
Regarding entry in the first mitosis, we observed that all three epigenetic markers were present: (1) H3S10P covered equally both parental genomes, (2) H3K9me3 strongly labeled the maternal genome and faintly stained the paternal one only at the very end of the one-cell stage, and (3) the intensity of H3K9meS10P clearly increased covering both parental genomes. Conversely, it is known that HP1β proteins preferentially localize within condensed inactive heterochromatin and that it dissociates from chromatin during mitosis (Hayakawa et al., 2003; Minc et al., 1999; Puschendorf et al., 2008). In fact, H3K9me3 and HP1 work together to propagate heterochromatin and cause gene silencing (Lachner et al., 2001), but the binding of HP1β to the methylated H3 tail is fully reversible and highly dynamic thereby supporting the rapid exchange of HP1β from heterochromatin (Fischle et al., 2005). It has been proposed in somatic cells that H3S10 phosphorylation prevents the binding of HP1 to the adjacent trimethylated lysine 9 residue of histone H3 (Fischle et al., 2003; Hirota et al., 2005).
Also separate observations of H3S10P staining versus H3K9me3 staining first gave the impression that the scenario in mouse embryos was in contradiction to the hypothesis for somatic cells. We then observed that both parental genomes present a strong signal H3K9me3S10P only upon the first mitosis, exactly when HP1 disappears (Puschendorf et al., 2008 and our own unpublished results). However, we do not know whether additional phosphorylation at serine 10 occurs on the already trimethylated lysine 9 histone H3 or vice versa. Experiments using ZM447439 (ZM), an inhibitor of Aurora kinases activity, which is required for phosphorylation of H3S10, showed that embryos lacking H3S10 phosphorylation almost completely did not cleave properly (Teperek-Tkacz et al., 2010 and our own unpublished results). Similarly, disruption of the two mouse Suv39h histone methyl transferases (HMTases) that abolish H3-lysine 9 methylation of constitutive heterochromatin induces gestation death or postnatal growth delay (Peters et al., 2001; Peters et al., 2002). Therefore, both phosphorylation at serine 10 and trimethylation on lysine 9 might be involved in embryonic chromosome condensation.
At the two-cell stage, heterochromatin undergoes massive distribution changes, moving from the rings surrounding the NPBs toward the nuclear periphery to form new heterochromatin domains, the chromocenters (Martin et al., 2006a; Merico et al. 2007). However, in cloned embryos, it appears that the heterochromatin markers H3K9me3 and HP1β have a distinct behavior. In early two-cell cloned embryos, the two markers were observed in rings around the NPBs, a characteristic of a normal early two-cell stage embryo, but also in numerous foci within the nucleoplasm (Martin et al., 2006a; Merico et al. 2007; this study). These foci most probably correspond to the remains of unremodeled heterochromatin clumps inherited from the donor cells. On the other hand, we observed that both H3S10P and H3K9me3S10P label only pericentromeric heterochromatin rings around the NPBs in the early two-cell stage, following the same nuclear movements in fertilized and cloned embryos. Therefore, it seems that the most important restructuring events occur during the first-cell stage, with relocalization of remodeled pericentric heterochromatin toward the NPBs, and that cloned embryos do not undergo further reprogramming at the two-cell stage. There have been few studies addressing the importance of genome reorganization after nuclear transfer. In these studies, inhibitors of specific epigenetic modifications like DNA methylation and histone acetylation were applied in an attempt to improve cloning efficiency by improving chromatin remodeling (Bui et al., 2010; Maalouf et al., 2009; Wang et al., 2007; Yamagata et al., 2007). It has been shown that the use of trichostatin A (TSA), an inhibitor of deacetylases, which is known to increase the acetylation levels in somatic cells and embryos, improves cloned mouse development (Wang et al., 2007). Moreover, culturing reconstructed embryos in the presence of this drug improved chromatin reorganization. The centromeric and pericentromeric heterochromatin pattern from the TSA-treated cloned embryos resembled more the spatial distribution seen in fertilized ones, and a lower amount of embryos displayed irregular heterochromatin clusters not associated to the NPBs (Maalouf et al., 2009).
Therefore, rectifying reprogramming of these epigenetic modifications at an early stage may be a strategy to improve cloning efficiency (Shao et al., 2009). Because H3S10P and H3K9me3S10P seem to be good markers to trace remodeled pericentromeric heterochromatin after nuclear transfer, we speculate that, by the use of specific drugs, we could increase the levels of this histone H3 phosphorylation, and somehow heterochromatin remodeling could be enhanced. Caffeine, a protein phosphatase inhibitor, has, for example, been used to treat oocytes before nuclear transfer. This treatment increased the frequency of PCC as well as the development of cloned sheep embryos (Choi et al., 2010; Lee and Campbell, 2008). These authors believe that this increases the removal of chromatin-bound proteins, thus allowing the access of oocyte-derived factors involved in the reprogramming of the somatic DNA. The same principle could also be applied in regard to histone H3 phosphorylation. Treating cloned embryos with caffeine would in all probability raise the levels of the Aurora kinase responsible for H3S10 phosphorylation, making the chromatin more accessible to remodeling factors and thus facilitating even more heterochromatin remodeling.
Footnotes
Acknowledgments
We are grateful to Michael Jeanblanc for his help with the experiments on H3K9me3 and image acquisitions. We would also like to thank the platform MIMA2 (Microscopie et Imagerie des Microorganismes, Animaux et Elements) for confocal microscopy and IERP for animal care. The present work was supported by INRA « Jeune Equipe » funding and the European CLONET (MRTN-CT-2006-035468) grant. K.M. also obtained support from the Fondation pour la Recherche Médicale (FRM).
Author Disclosure Statement
The authors declare that no conflicting financial interests exist.