
Abstract
Abstract
This study investigated the effects of serum-starvation, total confluence, and roscovitine treatment on cell-cycle synchronization of buffalo ear skin fibroblasts to the G0/G1stage and on the developmental competence of cloned embryos. Serum starvation of total confluence cultures for 24 h had a higher (p<0.05) proportion of cells at G0/G1stage (94.4%) compared with serum starved cyclic and nonstarved confluent cultures (76.8 and 86.0%, respectively), whereas differences between cyclic cells with or without serum starvation were not significant. The proportion of cells at G0/G1 was higher (p<0.05) with 20 and 30 μM roscovitine treatment than that with 10 μM (94.4, 96.4, and 86.6%, respectively), which was similar to that for total confluence (86.0%). MTT assay showed that cell viability decreased as dose of roscovitine increased. The blastocyst rate was significantly higher (p<0.05) when nuclear transfer embryos were reconstructed using donors cells from total confluence, confluence serum starved, and roscovitine-treated (20 and 30 μM) groups (48.8, 48.9, 57.9, and 62.9%, respectively) compared to nontreated cyclic cells (20.2%). However, the cleavage rate and total cell number of cloned embryos were similar for all the groups. The number of ICM cells was improved by 30 μM roscovitine treatment (45.25±2.34). The cryosurvival rate of blastocysts derived from cells synchronized with 20 or 30 μM roscovitine was higher compared to that for total confluence group (33.6, 37.8 vs. 23.8%). In conclusion, treatment with 30 μM roscovitine is optimal for harvesting G0/G1stage cells for producing high quality cloned buffalo embryos, and that it is better than serum-starvation or total confluence for cell synchronization.
Introduction
Buffalo cloning is still an inefficient process and our previous experiments in buffalo designed to compare the effects of in vitro culture conditions (Shah et al., 2009), source of donor nucleus (George et al., 2011; Selokar et al., 2011; Shah et al., 2008) and cytoplasmic volume (Panda et al., 2011) on overall efficiency of in vitro production of cloned embryos and their subsequent in vivo survival have shed new light on the cloning process in buffalo. It appears that the coordination between the cell-cycle stage of the donor nucleus and the recipient oocyte cytoplasm is essential for successful development of NT embryos for this species. However, there exists very limited information about cycle synchronization of buffalo cells and only the total confluence and serum starvation methods have been used for synchronization of donor cells. The cloning efficiency could improve if the method of cell cycle synchronization is adequate and appropriate. Therefore, the aim of the present study was to investigate the efficacy of various synchronization methods such as total confluence (TC), serum starvation (SS), combination of TC with SS (TCSS), and chemical synchronizer roscovitine (ROS) for synchronization of cultured buffalo fibroblasts, toward the general goal of improving the cloning efficiency in this species.
Materials and Methods
Unless stated otherwise, chemicals and growth media were purchased from Sigma (St. Louis, MO, USA), and antibodies for immunocytochemistry were obtained from Millipore (Billerica, MA, USA). All incubations were done in CO2 incubators (5% CO2 in air and maximum humidity) maintained at 38.5°C for for oocytes and embryos and 37°C for somatic cells. Experimental design of the present study has been shown in Figure 1.
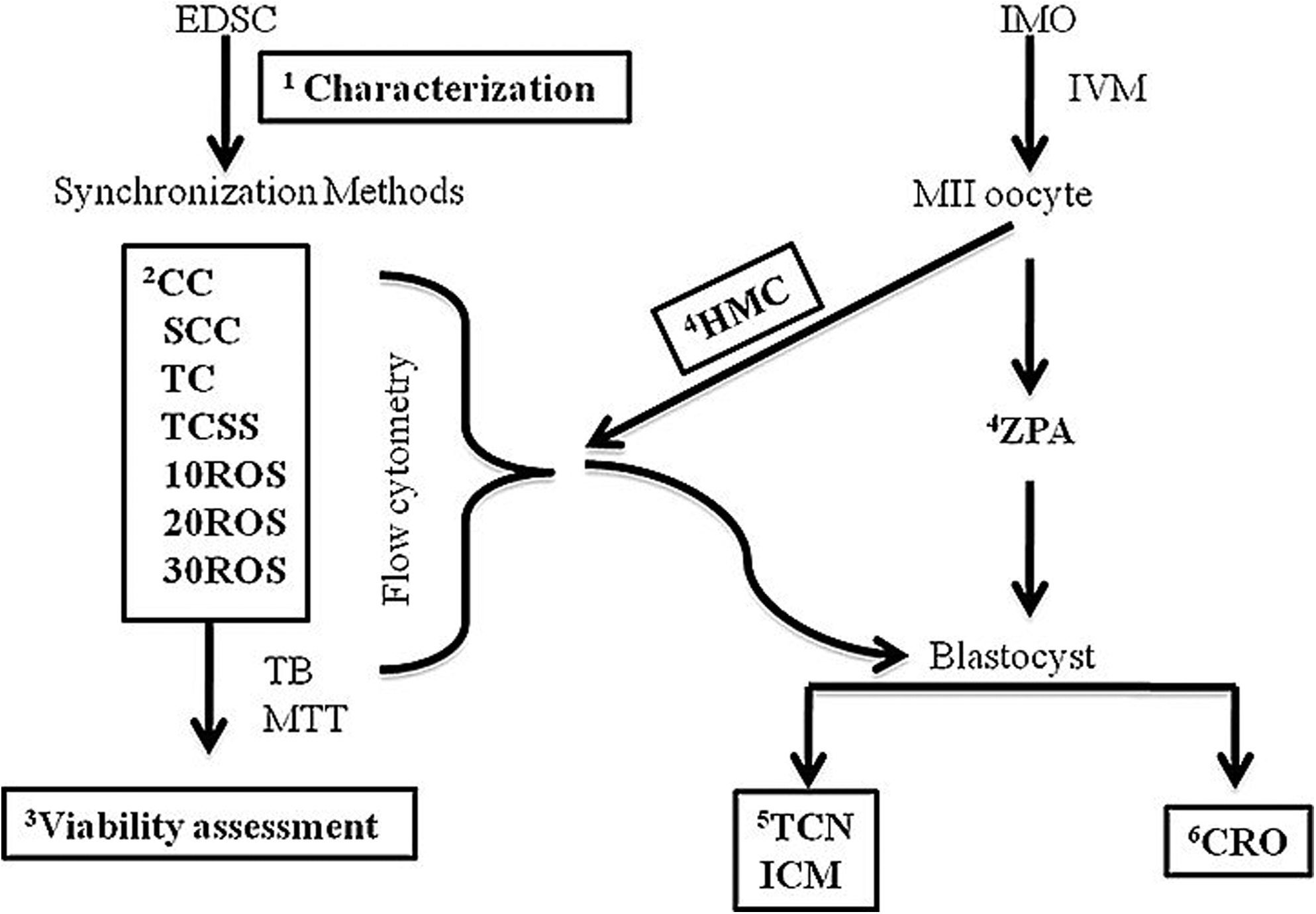
Experimental design. Numbers 1 to 6 represent the sequence of experiments during the study; EDSC: ear-derived somatic cells; CC (cyclic culture): cells cultured at 60–70% confluence; SCC: cyclic culture with serum starvation; TC: cells cultured to total confluence and then cultured for an additional 48–72 h for inducing contact inhibition; TCSS: total confluence+serum starvation; 10/20/30ROS: respective concentration of Roscovitine in μM; IMO: immature oocyte; HMC: hand-made cloning; ZPA: zona-free parthenogenesis as internal control; TB: trypan blue staining for cell viability; MTT: MTT assay for cell cytotoxicity; TCN: total cell number; ICM: inner cell mass and CRO: cryopreservation.
Isolation, culture, and characterization of buffalo skin cells
Buffalo cells were isolated from ear skin biopsies of fetus (60–90 days) and cultured as reported earlier (Shah et al., 2009). For immunocytochemical characterization, cells (2000/well) were cultured in 96-well plates and grown to 60–70% confluence. The cells were fixed for 30 min in paraformaldehyde (PF, 4% in DPBS with Ca2+ and Mg2+), and permeabilized in 1% Triton X-100 for 30 min. After three rinses with DPBS for 2 min each the cells were blocked with 4% (v/v) goat serum diluted in DPBS. The cells were rinsed again with DPBS, incubated with blocking solution containing primary mouse anticytokeratin (1:50) or antivimentin (1:100) for at least 1 h, rinsed three times with DPBS, and incubated with secondary goat antimouse IgG conjugated with FITC (1:100) for 1 h. The cells were rinsed and incubated in 10 μg/mL of Hoechst 33342 for 10 min to stain the nuclei and were then examined by epifluorescence microscopy (Diaphot; Nikon, Tokyo, Japan). Positive controls to test the efficiency of staining protocol were from each respective cell culture and were labeled with mouse antitubulin (1:100), whereas the negative controls were incubated with only the secondary antimouse FITC-conjugated antibody but not with respective primary antibodies.
Treatment of cells for cell cycle synchronization
The cells were seeded in six-well plates (1×105cells/well) and grown to 60–70% confluence to obtain cultures growing in the logarithmic phase, after which these were subjected to following treatments: (1) CC (cyclic culture): cells cultured at 60–70% confluence; (2) TC: cells cultured to total confluence and then cultured for an additional 48–72 h for inducing contact inhibition; (3) SCC: cells cultured at 60–70% confluence with serum starvation carried out by culturing the cells for 24 h in DMEM+0.5% fetal bovine serum (FBS); (4) TCSS: total confluence+serum starvation; (5) Roscovitine treatments: cyclic cells (60–70% confluence) were treated with roscovitine (10, 20, or 30 μM) for 24 h. In all the above experiments, the cells were cultured under similar culture conditions and were at passage number 6–10 during the study.
Determination of cell cycle stages by flow cytometry
For checking the efficiency of synchronization, the cells were analyzed through flow cytometry by propidium iodide staining as described previously (Riccardi et al., 2006) with some slight modifications. Briefly, the treated fibroblast cells were harvested by trypsinization and were fixed in chilled 70% ethanol at −20°C overnight. These were then incubated with DNA staining solution (100 μg/mL RNase, 50 μg/mL propidium iodide, and 1% Triton X-100) for 30 min. Subsequently, the cells were transferred to DPBS, after which flow cytometric analysis of cell cycle was done by using 488-nm laser line for excitation in a BD FACSCalibur (Becton-Dickinson, Rutherford, NJ, USA). For each cell treatment, 20,000 cells were analyzed, and the proportion of cells in G0/G1, S, and G2/M stage was estimated using the CellQuest cell cycle analysis program (Becton-Dickinson) with the same algorithm in all the treated samples. At least 3 independent flow cytometry experiments were performed for each synchronization method.
Assessment of cell viability/cytotoxicity by dye exclusion and MTT methods
For the determination of cell viability and cytotoxicity, trypan blue and MTT [3-(4, 5-di-methylthiazol-2-yl) 2, 5-diphenyl tetrazolium bromide] assays were conducted for treatment groups. To determine roscovitine-induced cytotoxicity, the treated cells were incubated with 5 mg/mL MTT at 37°C for 2 h, after which DMSO was added to the culture medium (1:1, by volume) and the reactants mixed by pipetting until the formazan was completely dissolved (Supplementary Fig. 1; Supplementary Data are available online at http:www.liebertonline.com/cell). The optical density of formazan was measured at 570 nm using a multiscan ascent plate reader. All analyses were performed in at least three independent replicate cultures. Absorbance ratio of treated to nontreated control cells was calculated and presented as relative cell viability.
Cloned embryo production from cells subjected to different synchronization procedures
Donor cell preparation was performed according to the method described previously (Shah et al., 2008), except that in the present study, the cells were synchronized by different methods before being used as karyoplasts. A total of 12 repetitions were carried out. However, not all treatments were included in each repetition. Preparation of recipient oocytes (maturation, cumulus/zona removal, and manual enucleation) and fusion, activation, and culture of NT embryos were performed according to methods described previously (Shah et al., 2008), with the minor modification such as increasing the holding time between electrofusion and activation from 4 to 6 h, and using the calcium inophore at a lower concentration (2 μM) for the activation of NT oocytes. Parthenogenetic activation and culture of zona-free oocytes were carried out just as for NT oocytes.
Assessment of embryo development and determination of blastocyst cell number
Cleavage and blastocyst rate were recorded on day 8 of IVC for assessing embryo development. For examining the health of the embryos, the cell number of trophectoderm (TE) and inner cell mass (ICM) of day 8 blastocysts was determined by differential staining as described by Thouas et al. (2001) with some modifications. Briefly, day 8 blastocysts were incubated in solution I (5 μg/mL Hoechst 33342 in DPBS) for 40 min, immediately transferred to solution II (0.4% Triton X-100 in DPBS) for 1 min, and finally transferred to solution III (25 μg/mL propidium iodide in DPBS) for 30–40 sec. Stained blastocysts were then washed with DPBS and were mounted on a glass microscope slide in a 3-μL droplet of glycerol and were flattened with a coverslip. Cell counting was performed from digital images obtained on an epifluorescence microscope (Diaphot; Nikon, Tokyo, Japan) fitted with an ultraviolet lamp and an excitation filter (460 nm) that allowed the visualization of the red and blue fluorochromes simultaneously (Supplementary Fig. 1).
Vitrification of cloned blastocysts generated through roscovitine-treated donor cells
The blastocysts produced were transferred to equilibration solution (ES) consisting of 7.5% ethylene glycol (EG) and 7.5% dimethylsulfoxide (DMSO) in T20 (TCM-119 containing 20% FBS) at 38.5°C for 15–20 min. After an initial shrinkage, the blastocysts regained their original volume. Four to six blastocysts were then transferred to vitrification solution (VS) consisting of 15% EG, 15% DMSO, and 0.5 M sucrose dissolved in T20. After incubation for 20–30 sec, the blastocysts were loaded into French ministraw (0.25 mL) with air space on both sides of embryos and were plunged into liquid nitrogen. The process from exposure in VS to plunging was completed within 1 min. For thawing, the straws were removed from liquid nitrogen and kept in air for 30 sec, after which these were plunged into warm water (39°C) for 40 sec before expelling the embryos in dilution solution I (1.0 M sucrose dissolved in T20) for 1 min and then in dilution solution II (0.5 M sucrose dissolved in T20) for 3 min. Subsequently, the blastocysts were incubated twice for 5 min in the washing solution (T20). Survival of vitrified blastocysts was determined according to reexpansion rates after 18–24 h of culture in RVCL medium (K-RVCL-50, Cook®, Australia) supplemented with 1% fatty acid-free bovine serum albumin (BSA).
Statistical analysis
The percentage data were analyzed using SYSTAT 7.0 (SPSS Inc.,Chicago, IL, USA) after arcsine transformation. Differences between means were analyzed by one-way ANOVA followed by Fisher's LSD test for percentage data of cell cycle stages, viability percentages, embryonic developmental stages,and cryosurvival rates. Differences were considered to be significant at p<0.05.
Results
Characterization of somatic cells isolated from ear tissue
The cells established for analysis of their cell cycle by flow cytometry were characterized by cell type-specific markers. To determine the type of somatic cells obtained from fetal ear samples, the cells were stained for examining the presence of cytokeratin and vimentin. Ear tissue-derived cells were vimentin positive but cytokeratin negative (Fig. 2) demonstrating that they were fibroblast cells.

Immunofluorescence analysis of ear-derived somatic cells at passages 12. (
Cell cycle synchronization of donor cells by various methods
To evaluate the effect of total confluence, serum starvation, and roscovitine treatment on the donor cell cycle stage, DNA content was measured by flow cytometric analysis, and the relative percentage of the proportions of cells in the G0/G1 (2C DNA content), S (2C–4C), and G2/M (4C) stages was calculated. Roscovitine treatment was found to improve the cell cycle synchrony. A higher (p<0.05) proportion of cells were synchronized in the G0/G1 stage of the cycle and consequently a lower proportion of cells were in the S and G2/M stage in the roscovitine groups (20 and 30 μM) compared with cyclic control, serum starvation and total confluence groups (Table 1). However, a combination of total confluence with serum starvation improved the synchronization of cells in G0/G1 stage, bringing it at par with roscovitine treatment.
Nonconfluent cycling fibroblasts cultured in DMEM+10% FBS for 24 h.
Nonconfluent fibroblasts cultured in DMEM+0.5% FBS for 24 h.
Confluent fibroblasts cultured in DMEM+10% FBS for 48–72 h.
Confluent fibroblasts cultured in DMEM+0.5% FBS for 24 h.
Roscovitine treated, nonconfluent fibroblasts cultured in DMEM+10% FBS for 24 h.
Data from three trials. Values are mean±SEM. Values with different superscripts within the same column differ significantly (p<0.05).
Assessment of cell viability/cytotoxicity
None of the synchronization methods significantly affected the cell viability, which was ∼90%, as indicated by trypan blue dye exclusion test (Fig. 3). However, MTT cytoxicity assay showed that the population of viable cells decreased as the dose of roscovitine increased (Fig. 3).

Effect of synchronization method on cell viability. (
Embryonic development of oocytes subjected to HMC after synchronization
Table 2 summarizes the embryonic development resulting from NT experiments. Donor cells that were synchronized into quiescence (G0/G1) stage of cell cycle by roscovitine (30 μM) treatment gave significantly higher (p<0.05) blastocyst rate than when these were subjected to TC or TCSS, which in turn, was higher (p<0.05) than that for cyclic control and serum starvation groups, although the cleavage rate was similar for all the treatment groups. Also, the embryos reconstructed with roscovitine-treated cells (30 μM) had a higher (p<0.05) number of ICM cells at the blastocyst stage than did the embryos reconstructed with cells synchronized with others methods (Table 2). However, the total cell number of blastocysts was not affected by the synchronization method used.
Data from 12 trials.
Values are mean ± SEM.
Values with different superscripts within the same column are significantly different (p < 0.05).
Parthenogenetic embryos were considered as internal control for activation protocol and oocyte quality.
For determining the number of cells in TCN and ICM, 20–50 blastocysts were analyzed by differential staining.
Cryosurvival of blastocysts derived from roscovitine-treated cells
The cryosurvival rate of vitrified-thawed cloned blastocysts derived from cells treated with 30 μM roscovitine was significantly higher (p<0.05) than that of blastocysts derived from cells treated with 20 μM roscovitine, which in turn, was higher (p<0.05) than that for untreated controls taken from total confluent group (Fig. 4).
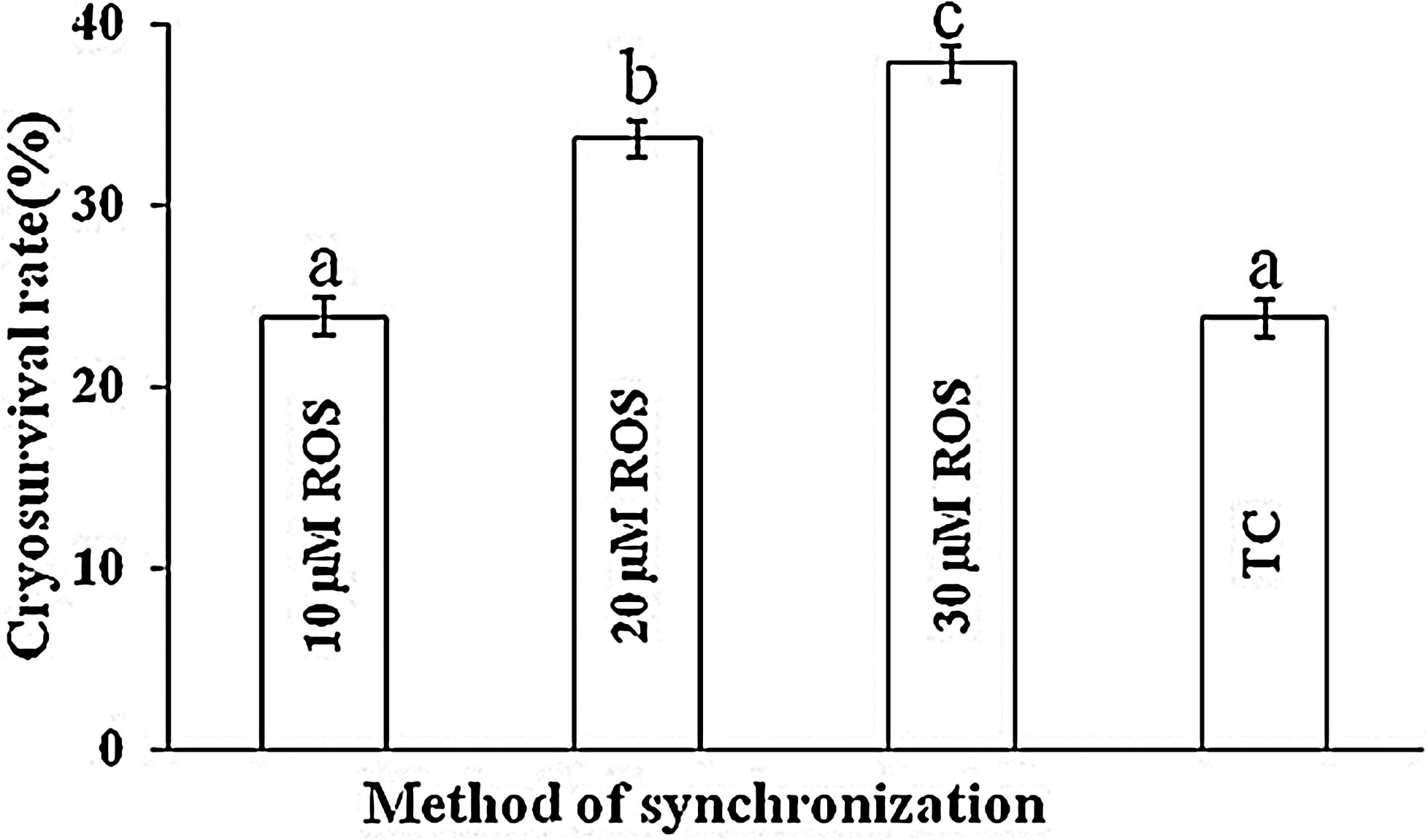
Cryosurvival of vitrified-thawed HMC buffalo blastocysts derived from roscovitine-treated donor cells and cells derived from total confluent (TC) group (control). Values are mean±SEM. Bars with different superscripts differ significantly (p<0.05). Data from four trials. A minimum 20 blastocysts were vitrified from each group and were kept in liquid nitrogen for 15–20 days before thawing. Normal reexpanded blastocysts were used for embryo transfer but data has been not shown because of less number of transfers.
Discussion
In the present study, we have analyzed various methods for the synchronization of cell cycle of buffalo ear fibroblasts. Cell cycle coordination between nucleus donor and recipient is considered to be a crucial factor for successful cloning for maintenance of the correct ploidy of embryos (Campbell et al., 1996). Hence, donor nuclei must be in G0/G1 stage when transferred to nonactivated oocytes arrested at the metaphase II stage. Data from the present study clearly indicates that ear skin fibroblasts can be synchronized effectively at G0/G1stage, which in turn, can successfully improve the in vitro developmental competence of cloned buffalo embryos. Most of the cells (>90%) were synchronized at the G0/G1stage when treated with high dose of roscovitine (20 or 30 μM) or when subjected to a combination of total confluence with serum starvation compared to around 70% cells found to at the G0/G1 stage in control cells of cyclic culture. In NT experiments, a high rate of blastocyst production (62.9%) was obtained using 30 μM roscovitine-synchronized cells. Also, these embryos had a higher number of ICM cells and improved survival after cryopreservation.
Since the birth of “Dolly,” serum starvation has perhaps been the most frequently used method for cell cycle synchronization in SCNT research (Wilmut et al., 2007). It has long been known to result in withdrawal of growth factors and other related components essentially required the cultured cells (Chesne et al., 2002). As a result, the cells undergo a rapid exit from the cell cycle and enter the G1 stage, which is characterized by low metabolic activities (Iyer et al., 1999; Kues et al., 2000). Alternately, the cultured cells synchronize at the G1 stage, a nondividing state, once they come into contact with each other (Suzuki et al., 2000). Although little is known about the molecular mechanisms involved in this phenomenon, contact inhibition/total confluence is also a well-known and commonly used method for cell cycle synchronization. Another approach for cell cycle synchronization is the use of chemicals that control check points of cell cycle progression. Cell cycle progression is regulated by serine/threonine kinases, termed cyclin-dependent kinases (CDKs), the activities of which oscillate during the cell cycle. CDKs are associated with the positive coactivators, cyclins, and the negative regulators, CDK inhibitors (Pines, 1995; Sherr and Roberts, 1995). In mammalian cells, cyclin E-CDK2 and cyclin A-CDK2 complex regulates the progression of G0/G1 to S phase (Johnson and Walker, 1999; Suzuki et al., 2000). Roscovitine is a CDK2 inhibitor that reduces the effects of cyclins E and A. As a result, roscovitine treatment has been shown to effectively induce cell cycle synchronization at the G0/G1 stage in various species (Gibbons et al., 2002; Hinrichs et al., 2007).
Effective synchronization of donor cells in G0/G1 stage has been achieved by serum starvation for 4 days in sheep (Wilmut et al., 1997), 5–10 days in rabbit (Zhou et al., 2006) and 5 days in cattle (Kubota et al., 2000). In the present study, a combination of total confluence with serum starvation for 24 h synchronized >90% cell population in G1/G0 stage, whereas Sun et al. (2008) obtained >90% cattle fibroblasts cells to be in G0/G1stage after 3–5 days of serum starvation. The differences in results may be due to species differences, individual variation, cell types, and treatment methods. Serum starvation of cyclic cells for 24 h resulted in an increase in the proportion of cells at G2/M stage (16.3%) and a decrease in proportion of cells at S (15.8%) compared to that of nonstarved cyclic cells. However, this did not significantly increase the proportion of synchronized fibroblasts at G0/G1stage (76.8%), which is contradictory to the report of Miranda et al. (2009), who observed an improvement in G0/G1 stage synchrony after 24 h of serum starvation (82%). Besides species differences, the variation between our results and those of these authors could be due to different source of the cells because we used cells of fetal origin, whereas these authors used adult cells. Thus, our results and those of Shi et al. (2007) indicate that 24 h of serum starvation of cyclic cells is not effective for cell cycle synchronization to the G0/G1 stage in buffalo. Culture to total confluence is another strategy used by us to move cells into G0/G1 stage. In our study, 85% of cells that were subjected to total confluence were found to be in the G0/G1 stage (Table 1), which agrees well with earlier reports in porcine (Boquest et al., 1999) and bovine fibroblasts (Cho et al. 2005). We also tested the response of buffalo fibroblasts to the cell cycle inhibitor roscovitine, which has been shown to arrest cultured fetal fibroblast cells from cattle in G1/G0 (Cho et al., 2005). Gibbons et al. (2002) reported that roscovitine-treated adult bovine granulosa cells were more efficiently synchronized in G0/G1stage of the cell cycle than serum-starved cells. Our results showed that the synchronization efficiency of roscovitine treatment was dose dependent and that it was higher than that of other methods such as serum starvation and total confluence.
In the present study, all the methods examined were able to induce cell cycle synchrony at the G1/G0 stage. However, the proportion of viable cells that each treatment method offered was of primary concern for the NT work. It is well known that serum starvation induces DNA fragmentation and apoptosis (Kues et al., 2000) and that this damage may be a cause of embryo/fetal loss and abortion in cloned embryos (Kato et al., 1998; Hill et al., 1999; Vignon et al., 1998). In present study, cell viability was determined by trypan blue dye exclusion method and colorimetric MTT assay. The latter has been shown to be useful for measuring the active viability of murine macrophages (Ferrari et al., 1990) and human monocytes (van de Loosdrecht et al., 1991) and NK/LAK cells (Heo et al., 1990; Hussain et al., 1993). The MTT assay, originally developed by Mossman (1983), is based on the reduction of MTT by the mitochondrial dehydrogenases of living cells (Supplementary Fig.1). On the basis of trypan blue test, the proportions of viable cells was found to be >90% in all the groups and was not affected by the method of synchronization. However, this test is indicative of immediate survival and does not always predict ultimate survival. Therefore, MTT assay was carried out to check the cytoxicity of roscovitine treatment also because we observed some morphological changes in the roscovitine-treated cells. We found that the proportion of viable cells decreased as dose of roscovitine increased. The differences in the results of MTT assay and trypan blue exclusion method could be due to the fact that viability detected by the former is based on the mitochondrial metabolic activity rather than on membrane integrity detected by the latter.
In our study, the use of cells synchronized by treatment with roscovitine (30 μM) as nuclear donors for SCNT improved both blastocyst rate (p<0.05) and the number of ICM cells of blastocyst (p<0.05) compared to when cells synchronized by other treatments were used. Nonetheless, the cleavage rate was similar for all the groups indicating that the early embryo development was perhaps not compromised because of the donor cell cycle stage prior to NT. Subsequent development was, however, significantly affected, as indicated by a blastocyst rate of 20% for cyclic cells compared to that of 62% for the cells synchronized by treatment with 30 μM roscovitine. However, our results also demonstrate that the blastocyst rate following roscovitine treatment was dose dependent and higher than that following serum starvation and total confluence treatments. Gibbons et al. (2002) reported that roscovitine-treated adult bovine granulosa cells had a lower blastocyst rate than serum starved cells, although the surviving calf efficiency was more in roscovitine-synchronized cells. A small number (n=15) of cloned blastocysts derived from roscovitine-treated cells was transferred nonsurgically to suitably synchronized recipients but no conclusions can be drawn due to the small number of transfers. The total cell number of cloned embryos was not affected by the method of synchronization used, although the ICM cell numbers were highest in blastocysts derived from cells treated with 30 μM roscovitine. Several reports have shown that the developmental competence of cloned embryos derived from donor cells at the G0 stage was higher than that of embryos derived from cells at the G1 stage (Kasinathan et al., 2001a, 2001b; Wells et al., 2003). Roscovitine induces cells into the G0 stage because it inhibits the activity of cyclins, rather than inducing a resting, nondividing state (G1). This could be the reason for improved blastocyst rate obtained in the present study. Further research is, however, needed to evaluate the effects of roscovitine treatment of donor cells on in vivo developmental competence of SCNT embryos in buffalo.
The cryosurvival rate of cloned blastocysts derived from roscovitine-treated cells after vitrification in straws was also examined. Embryo survival after freezing can be assessed by post thaw reexpansion of blastocysts (Agca et al., 1994; Ishimori et al., 1995; Kobayashi et al., 1994) and assessment of the hatching rate (Dinnyes et al., 1996, Hochi et al., 1996; Saha et al., 1996; Vajta et al., 1998). In the present study, the postthaw viability was accessed by reexpansion of blastocysts after culturing them in RVCL medium for 18–24 h (Supplementary Fig. 1). The cryosurvival rate obtained in the present study with blastocysts derived from cells subjected to total confluence was similar to that of 30% reported by Yang et al. (2010) for vitrified-thawed cloned blastocysts in buffalo but was lower that obtained after roscovitine treatment of cells.
In conclusion, we have shown that buffalo fibroblast cells can be synchronized at the G0/G1 stage using total confluence, serum starvation or roscovitine treatments. NT results showed that monitoring the cell-cycle stage distribution for each cell line and for each treatment before their use as donor cell is of great value. Among the different methods of cell cycle synchronization examined in the present study, roscovitine treatment gave the highest blastocyst rate and number of ICM cells/blastocyst. Previous reports on SCNT in the bovine (Gibbons et al., 2002) and equine (Hinrichs et al., 2007) species demonstrated that cell cycle synchronization using roscovitine reduced embryonic and fetal loss. Whether its application to buffalo cells prior to NT offers similar benefits needs to be examined.
Footnotes
Acknowledgments
The authors thank Dr. Anil Suri, Manoj Kumar, and Sweta for assistance in flow cytometry analysis of cell cycle. This work was supported by research grants from National Agriculture Innovative Project (1(5)/2007-NAIP-2) to S.K.Singla. N.L.S. is recipient of Canadian Commonwealth Fellowship study program.
Author Disclosure Statement
The authors declare that no conflicting financial interests exist.
References
Supplementary Material
Please find the following supplemental material available below.
For Open Access articles published under a Creative Commons License, all supplemental material carries the same license as the article it is associated with.
For non-Open Access articles published, all supplemental material carries a non-exclusive license, and permission requests for re-use of supplemental material or any part of supplemental material shall be sent directly to the copyright owner as specified in the copyright notice associated with the article.