
Abstract
Abstract
Novel stem cells expressing stage-specific embryonic antigen 3 (SSEA-3) reside among human dermal fibroblasts and are known as multilineage-differentiating stress-enduring (Muse) cells. They enhance the generation efficiency of induced pluripotent stem cells. However, Muse cells have only been found in humans. We aimed to isolate SSEA3-positive cells from terminally differentiated skin fibroblasts of adult goat and determine their pluripotency. Cell clusters from SSEA3+ populations possessed stem cell-like morphological features and normal karyotypes, were consistently positive for alkaline phosphatase, and expressed stem cell pluripotency markers. These SSEA3+ cells remained undifferentiated over eight passages in suspension culture and were able to differentiate into cells of all three germ layers in vitro and in vivo. Our combined findings suggest that a subset of adult stem cells expressing SSEA3 also exist among adult goat skin fibroblasts. We are the first to report that multipotent adult goat cells exist among terminally differentiated goat skin in suspension culture. Our results also provide a promising platform for generation of a transgenic goat, because the undifferentiated state of stem cells was thought to be more efficient as donor cells for somatic cell nuclear transfer.
Introduction
To date, skin stem cells have been isolated from various species, including mice (Toma et al., 2001), pigs (Dyce et al., 2004, 2006), and humans (Kuroda et al., 2010; Toma et al., 2005). These studies have showed that stem cells could be derived from the dermis, and these have been designated SKPs. Clones of individual stem cells growing in suspension can express pluripotency markers and actively proliferate in vitro after long-term culture. These stem cells can only differentiate into neural and mesodermal progeny. Recently, a report showed that multilineage-differentiating stress-enduring (Muse) cells expressing stage-specific embryonic antigen 3 (SSEA3) reside among human dermal fibroblasts (Kuroda et al., 2010). These Muse cells express pluripotency markers and differentiate into cells of the endodermal, ectodermal, and mesodermal germ layers in vitro and in vivo (Kuroda et al., 2010). Muse cells can enhance the generation efficiency of induced pluripotent stem cells (iPSCs), with Muse-iPSCs demonstrating a formation efficiency of 0.03%, 30-fold more efficiently than that for fibroblasts (approximately 0.001%) (Byrne et al., 2009; Wakao et al., 2011). These findings indicated that the SSEA3+ cells from skin fibroblasts can be more easily reprogrammed into iPSCs compared with cells that do not express SSEA3. However, to date, Muse cells have not been found in animal species other than humans.
Traditionally, isolation and expansion of undifferentiated pluripotent stem cells have been based mostly on methods developed for mouse embryonic stem cells (mESCs), such as using feeder layers of inactivated mouse embryonic fibroblasts (MEFs) (Thomson et al., 1998). However, these approaches risk microbiological contamination, require separation of feeder cells from the cell type of interest, are expensive, and are prone to variability (Ährlund-Richter et al., 2009). Findings from a recent study suggest that suspension cultures can increase the number of high-quality undifferentiated pluripotent stem cells (Chen and Pei 2012). In contrast to adherent culture, stem cells in suspension cultures are more likely to maintain an undifferentiated phenotype and pluripotency (Amit et al., 2010, 2011; Fluri et al., 2012; Zweigerdt et al., 2011).
We hypothesized that SSEA3+ stem cells also exist among skin fibroblasts of adult goats. We attempted to isolate and characterize SSEA3+ stem cells from goat skin fibroblasts using suspension culture.
Materials and Methods
Reagents
Unless otherwise stated, all chemicals and reagents were purchased from Sigma Chemical Company (St. Louis, MO, USA), cell culture media were obtained from Gibco (Grand Island, NY, USA), and all antibodies were from Abcam Inc. (Cambridge, MA, USA). Disposable, sterile plasticware was purchased from Corning, and filters were from Millipore (India Pvt. Ltd. India). The same batch of fetal bovine serum (FBS) was used throughout the study.
Animals and tissues
All experiments were approved by the Animal Care Commission of the College of Veterinary Medicine, Northwest A&F University. Adult Saanen dairy goats were provided by Yangling Keyuan Cloning Co., Ltd., China. We biopsied skin tissue (approximately 0.5 cm×0.6 cm) from the ears of adult goats.
Cell culture
The skin tissue biopsies were washed three times with Ca2+/Mg2+-free phosphate-buffered saline (PBS). After careful removal of conjunctive tissue and subcutaneous fat, the sample was sterilized with 70% (vol/vol) ethanol, rinsed in PBS, and minced into small pieces (approximately 1×2×3 mm) using a sharp scalpel; pieces were then seeded in 60-mm cell culture dishes. Cells were maintained in Dulbecco's modified Eagle medium (DMEM) containing 10% FBS, 2 mM
All cultured mESCs were maintained on MEF cells in high-glucose DMEM containing 15% FBS and 1000 IU/mL leukemia inhibitory factor (LIF), along with nonessential amino acids,
Stress induction assay
A previous study revealed that SSEA3+ stem cells can be enriched by treating them with trypsin over different incubation periods for different times (Kuroda et al., 2010). We explored whether stressing cells through poor nutrition, low serum levels, and trypsin treatment enriched the population of SSEA3+ stem cells. Fibroblasts at passage 3 (P3) were divided into groups according to culture conditions. Group I cells were cultured in serum-free medium for 2 days; group II cells were cultured in PBS for 2 days; and cells in groups III–VII were incubated with trypsin for 4, 8, 12, 16, and 20 h, respectively. Fibroblasts that were not subjected to any stressors were used as negative control. For trypsin treatment, approximately 5×105 cells were suspended in 5 mL of 0.25% trypsin solution and incubated at 37°C/5% CO2 in 60-mm culture dishes. Three fibroblast cell lines were used, and experiments on each line were replicated three times.
Suspension culture
When cells were subjected to stress, a large number of dead cells were produced, adversely affecting the survival and growth of viable cells. To eliminate this cellular debris, continuous vortexing and centrifuging was conducted. Cell viability was determined by Trypan Blue staining. Cells were transferred to ultra-low-attachment dishes (day 0) and maintained in DMEM containing 10% FBS at 37°C/5% CO2, and cell density was maintained at around <5000 cells/mL. A volume of culture medium equal to 20% of initial culture medium was added to the dish every 2 days. Clusters of cells were counted on day 7, and the cell cluster formation rates of adult fibroblasts under different stress conditions were evaluated.
Alkaline phosphatase staining
The cell clusters and mESCs cells derived from suspension culture were washed at last three times (5 min each wash) with PBS containing 0.2% polyvinyl alcohol (PVA). Staining was performed at room temperature in the dark using the 5-bromo-4-chloro-3′-indolyphosphate and nitro-blue tetrazolium (BCIP/NBT) Alkaline Phosphatase Color Development Kit (Beyotime, Jiangsu, PR China) according to the manufacturer's instructions.
Immunofluorescence staining
Cells were washed three times (5 min each) in PBS containing 0.2% PVA, and fixed in Immunol Staining Fix Solution (Beyotime, P0098) for 1 h. After three further washes, cells were permeabilized with 0.1% Triton X-100 in PBS for 30 min, then incubated with Immunol Staining Blocking Solution (Beyotime) at 4°C overnight. We incubated samples with specific antibodies as against the following markers: Nanog (1:100, ab21624), Oct4 (1:100, ab19857), Sox2 (1:100, ab97959), TRA-1-60 (1:100, ab16288), anti-stage-specific embryonic antigen 1 (SSEA1) (1:100, ab16285), anti-stage-specific embryonic antigen 3 (SSEA3) (1:100, MAB1434, R&D, Inc., Minneapolis, MN, USA), anti-stage-specific embryonic antigen 4 (SSEA4) (1:100, ab16287), CD105 (1:100, ab53318), CD34 (1:100, ab81289), CD271 (1:100, ab8877), Snail (1:100, ab82846), Slug (1:100, PA5-11921, Thermo, Cambridge, IL, USA), nestin (1:100, ab11306), neurofilament (1:100, ab7794), β-III tubulin (1:100, ab7751), alkaline phosphatase (ALP; 1:100, ab17272), and α-fetoprotein (1:100, ab3980).
Secondary antibodies we used were: Alexa Fluor 488-labeled goat anti-rabbit immunoglobulin G (IgG; 1:500; A0423, Beyotime), Alexa Fluor 488-labeled goat anti-mouse IgG (1:500; A0428, Beyotime), goat anti-rat IgG (1:500, sc3740, Santa Cruz Biotechnology, Inc., Santa Cruz, CA, USA), and fluorescein isothiocyanate (FITC)-labeled goat anti-mouse IgG (1:500; A0568, Beyotime).
Samples were incubated with primary antibodies diluted in Immunol Staining Primary Antibody Dilution Solution (Beyotime) overnight at 4°C. After washing three times, samples were incubated with secondary antibodies diluted in Immunol Staining Secondary Antibody Dilution Solution (Beyotime) for 2–3 h at room temperature in the dark. DNA was stained with 4′,6-diamidino-2-phenylindole (DAPI) (Beyotime) for 3 min. Negative controls were only stained with secondary antibodies.
Images were acquired with a Nikon Eclipse Ti-S microscope equipped with a 198 Nikon DS-Ri1 digital camera (Nikon, Tokyo, Japan) using the same laser intensity and detection sensitivity. Immunocytochemistry for Nanog, Oct4, Sox2, TRA-1-60, CD105, and SSEA3 in clusters was repeated at least five times with different samples used for each marker. For in vitro–differentiated SSEA3+ clusters, we repeated the immunocytochemistry staining for nestin, neurofilament, β-III tubulin, ALP, and α-fetoprotein three times on different samples.
Flow cytometry
For cell sorting, skin primary fibroblast cells were incubated with an antibody against SSEA3 (1:100, R&D Systems) for 30 min at 4°C. After washing three times in PBS, cells were incubated with phycoerythrin (PE)-conjugated goat anti-rat IgG (1:500, Santa Cruz) for 30 min at 4°C. Cells were washed three times and resuspended in 2 mL of ice-cold PBS with 2% FBS, then immediately analyzed and sorted on an EPICS XL-MCL cell sorter (Beckman Coulter) using a low stream speed to ensure a high level of cell survival and to obtain the highest purity of sorted cells.
SSEA3+ cells were also sorted from skin fibroblast cells using long-term trypsin incubation (LTT). We analyzed SSEA3+ cells for the expression of cell-surface antigens such as CD105, CD34, CD271, Snail, and Slug. In addition, we also analyzed the expression of Nanog and Oct4 among purified SSEA3+ cells, with goat fibroblast cells used as negative controls. The SSEA3+ percentage of the purified SSEA3− cells by fluorescence-activated cell sorting (FACS) was also analyzed. Isotype-specific FITC- or PE-conjugated goat anti-mouse IgG, goat anti-rabbit IgG, and goat anti-rat IgG antibodies were also used as negative controls. Gating strategies and data analysis were performed with EXPO32 ADC analysis software (Beckman Coulter).
Karyotyping
Cell clusters at P2, P5, and P8 were subjected to chromosomal analysis. According to a previously published protocol (Nguyen and Pera, 2008), cells were treated with 0.03–0.05 μg/mL colchicine for 3–6 h. Harvested cells were incubated in 0.07 mM KCl to enhance swelling at 37°C for 30 min. After centrifugation, cells were fixed in Carnoy's solution (methanol:glacial acetic acid, 3:1) at 37°C for 20–30 min, which we repeated twice. After the cell suspension was dropped on a clean precooled glass slide, the metaphase chromosomes were stained with 2% Giemsa stain for 5 min and examined at 1000× magnification under oil.
Reverse transcription polymerase chain reaction
Goat fibroblast cells (approximately 100,000 cells/well in a six-well plate), SSEA3+ clusters (first and fifth generations), and SSEA3− cells from goat fibroblast cells, and in vitro differentiated cells in vitro from SSEA3+ clusters were subjected to reverse transcription polymerase chain reaction (RT-PCR). Total RNA was extracted from cells and purified using an RNAprep pure Micro Kit (Qiagen, Mississauga, ON, Canada). First-strand cDNA was generated using a PrimeScript® RT reagent Kit Perfect Real Time (TaKaRa, Japan) according to the manufacturer's instructions. The PCR reactions were conducted using Ex Taq DNA polymerase (TaKaRa, Japan). Goat liver tissue was used as positive control for α-fetoprotein. The following oligonucleotide primers were designed and used: 5′-GAA GCT GGA CAA GGA GAA GCT-3′ and 5′-CAT GCT CTC CAG GTT GCC TC-3′ (Oct4); 5′-GAA AAA CCA AGA CGC TCA T-3′ and 5′-CTG GAG TGG GAA GAA GAG GT-3′ (Sox2); 5′-ATG CCT GAA GAA AGT TAC GC-3′ and 5′-AGG CTG TAT GTT GAG AGG GT-3′ (Nanog); 5′-CGG TGC CCA TCT ATG AGG-3′ and 5′-GAT GGT GAT GAC CTG CCC-3′ (β-actin); 5′-TCT TCT TGT TTA AAA TCC TAA CCT-3′ and 5′-CTC AAC AGT TCT ATC TCT TCT TCA-3′ (MAP-2); 5′-CAC TGG AGG GCA CCA AGA AGG G-3′ and 5′-CAC GAG GTC CTT GTC GAT GTC C-3′ (osteonectin); and, 5′-GGG CTT TCC TCG TGT AAC C-3′ and 5′-CCA CCC TTC CAG TTT CCA G-3′ (α-fetoprotein).
In vitro differentiation
Individual clusters were picked up with a glass micropipette on day 7 and transferred to gelatin-coated dishes to expand cells. Expanded cells were separated into three fractions that were subjected to three different induction conditions.
For neural induction, expanded cells at a density of 1.0×104 cells/cm2 were cultured in an ultra-low-attachment dish for 7–10 days in Neurobasal medium supplemented with B-27 supplement, 30 ng/mL basic fibroblast growth factor (bFGF; PeproTech Inc., Rocky Hill, NJ, USA), and 30 ng/mL epidermal growth factor (EGF; PeproTech) to induce sphere formation (Hermann et al., 2004). Some neural stem cell spheres were subjected to immunofluorescent staining for nestin, while the remaining spheres continued to be induced as previously described (Ambasudhan et al., 2011; Przyborski et al., 2008; Woodbury et al., 2000). Neurospheres were transferred onto gelatin-coated dishes and maintained in α-minimal essential medium (α-MEM) supplemented with 2% FBS, 25 ng/mL bFGF, and 25 ng/mL brain-derived neurotrophic factor (BDNF) for 21–30 days. Culture medium was changed every 3 days.
For osteocyte induction, the expanded cells were cultured for 14–21 days in DMEM supplemented with 10% FBS, 50 μg/mL VC (Sigma), 50 mM β-glycerophosphate, and 1 μM dexamethasone (Sigma) on gelatin-coated dishes with medium change every 2 days (Cheng et al., 1994; Rickard et al., 1994).
For hepatocyte induction, expanded cells were cultured on gelatin-coated dishes in DMEM containing 10% FBS, 10× insulin-transferrin-selenium (ITS), 10 nM dexamethasone (Sigma), 100 ng/mL hepatocyte growth factor (Sigma), and 50 ng/mL FGF-4 (Sigma) for 14–21 days (Chen et al., 2007; Ong et al., 2006; Zhou et al., 2007) with culture medium changed every 2 days.
Goat fibroblast cells cultured in induction medium were used as negative controls. The induced differentiated cells were subjected to RT-PCR analysis and immunocytochemistry for nestin, neurofilament, β-III tubulin, ALP, and α-fetoprotein.
Injection of SSEA3+ cells into immunodeficient mice
Approximately 5.0×106 SSEA3+ cells were harvested and injected into the hind limb muscles of nude mice. After 8 weeks, mice were killed and the resultant teratomas were harvested for histological analysis using Hematoxylin & Eosin (H&E) staining. Additionally, J1 mESCs and MEFs treated with mitomycin C were injected into mice for use as positive and negative controls, respectively.
Statistical analysis
All data were analyzed using SPSS 16.0 statistical software (IBM Corporation, Somers, NY, USA). Date were tested using one-way analysis of variance (ANOVA) and least significant difference (LSD) tests and reported as the mean±standard error of the mean (SEM). For all analyses, a p value less than 0.05 was considered significant.
Results
Enrichment and culture of skin stem cells
Three skin fibroblast cell lines were established for isolating of SSEA3+ skin stem cells (Fig. 1A). We explored whether stressful conditions [low serum and LTT] could enrich the population of putative multipotent stem cells. Surviving fibroblasts following stress treatments were suspended in ultra-low-attachment dishes at a density of 5000 cells/mL, forming cell clusters with diameters of 20–150 μm after 4 days subjected to the relevant condition (Fig. 1B). The rates of cluster formation were different among various groups (Table 1). Stress condition II produced large numbers of dead cells, which had toxic effects on the remaining viable cells. In our work, the formation rate of cell clusters for stress conditions I and III–VII were significantly higher than that for the control group. Of the six stress conditions, conditions IV and V showed the highest clone formation rate (respectively 11.54%±0.31% and 11.50%±0.85%, respectively), which was four-fold higher than that for the control group (2.68%±0.08%). LTT was able to enrich the number of cell clusters, and LTT for 8–12 h was most effective.
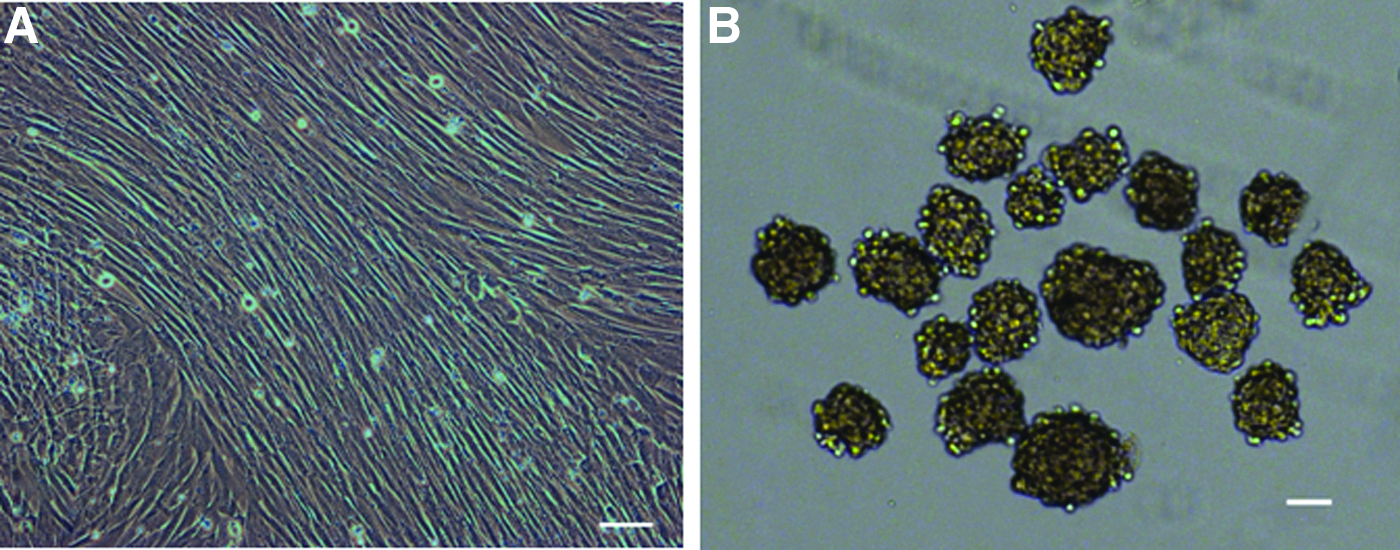
(
Note: I, 2 days nonserum-containing medium treatment; II, 2 days PBS buffer treatment; III, 4-h LTT treatment; IV, 8-h LTT treatment; V, 12-h LTT treatment; VI, 16-h LTT treatment; VII, 20-h LTT treatment. Fibroblast cells without stress treatment served as negative controls. After stress condition, the ratio of cell clusters formed was evaluated for 7 days of culture.
Average values of each group.
Values with different superscripts within a column are significantly different (p<0.05).
Characterization of cell clusters
Cell clusters larger than 25 μm were picked on day 4 and continued in suspension after limiting dilution. Until day 7, most cell clusters were morphologically similar to mESC colonies (Fig. 2A, B). Cell clusters larger than 25 μm were positive for AP staining (Fig. 2C, D). Immunofluorescence staining showed that the cell clusters were also positive for Nanog, Oct4, Sox2, TRA-1-60, CD105, and SSEA3 (Fig. 2E–L and O–P), but negative for SSEA1 and SSEA4 (Fig. 2M, N). These cell clusters also expressed pluripotent genes such as Oct4, Sox2, and Nanog (Fig. 2Q), which was validated by PT-PCR assays. Karyotype analysis verified that clusters maintained normal diploid karyotypes (Fig. 1R).
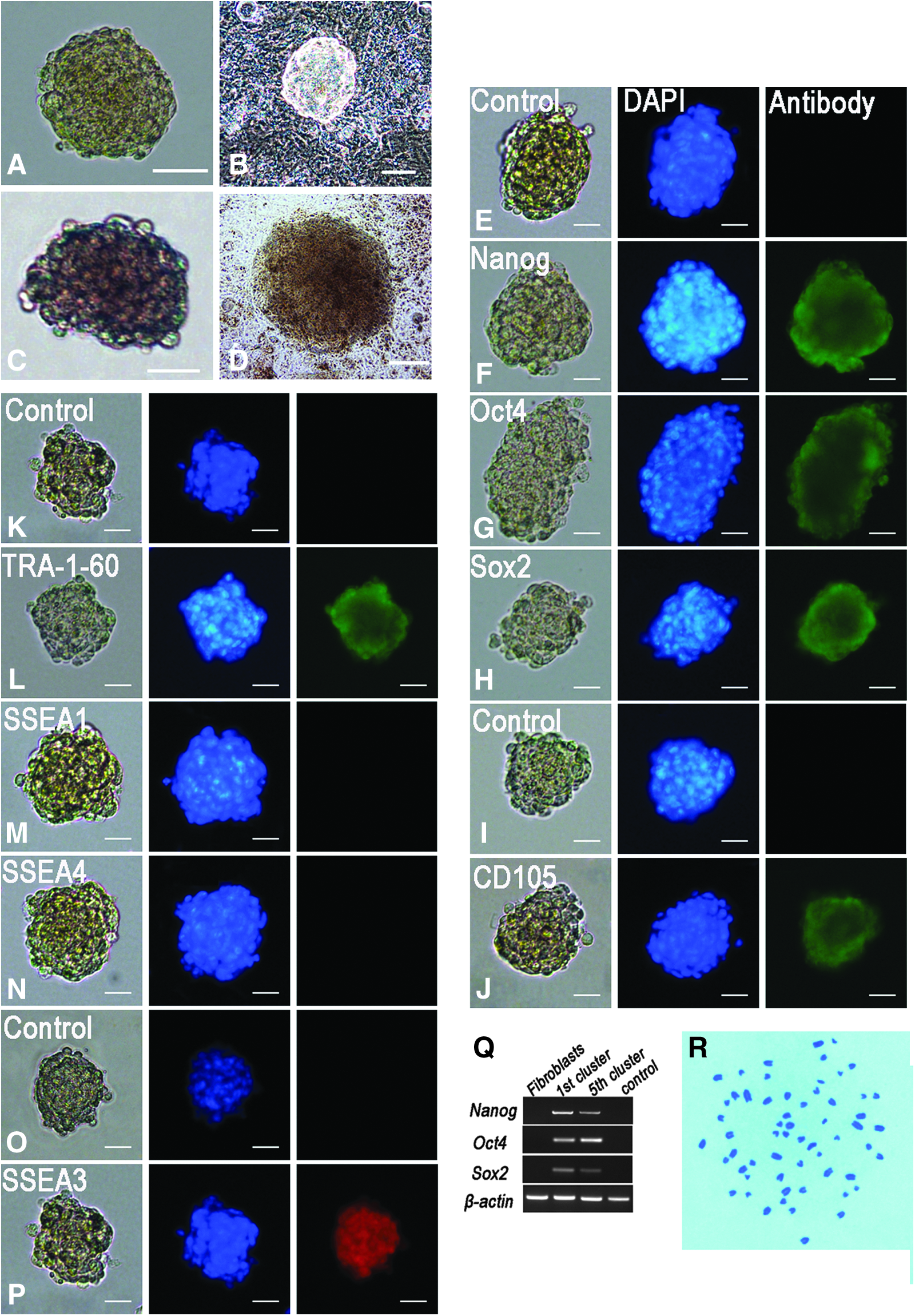
Characterization of SSEA3+ clusters. (
We also evaluated the expression of various markers in SSEA3+ clusters, such as CD146, CD271, Snail, and Slug. Immunofluorescence staining demonstrated that SSEA3+ cell clusters did not express these markers.
Self-renewal of SSEA3+ stem cells
When cell clusters larger than 25 μm in diameter were treated with 0.25% trypsin for 15 min, they could be separated into single cells. When these single cells were cultured in suspension, only a few cell clusters formed. Then we attempted to transfer single cell clusters to gelatin-coated dishes. Cells adhered to the culture surface and expanded from clusters. When clusters expanded to a single layer cell, cells were collected using a 5-min trypsin treatment and again subjected to suspension culture without LTT. We observed that 45.0%±6.3% of the cells could form clusters. However, when cells subjected to LTT for 8–12 h were grown in suspension, only 13.2%±1.8% of cells formed clusters. Thus, we repeated this culture cycle eight times and observed clusters forming with the same frequency at every cell generation. The eighth-generation clusters were still positive for ALP and for pluripotency markers, and lacked any abnormal karyotypes.
Flow cytometry
Untreated goat fibroblasts, and those subjected to LTT for 8–12 h were analyzed by flow cytometry. Approximately 3–4% of control goat fibroblasts were positive for SSEA3 (Fig. 3A), whereas about 13% of goat fibroblasts subjected to LTT for 8–12 h were SSEA3+ cells (Fig. 3B). To further investigate the pluripotency of these SSEA3+ cells, we conducted FACS analysis of the purified SSEA3+ cells using antibodies against Nanog and Oct4. We observed that few of the goat fibroblast cells expressed Oct4 and Nanog (Fig. 3C); however, approximately 80–90% of the purified SSEA3+ cells were positive for Nanog (86.6%) and Oct4 (83%; Fig. 3D). Examination of CD271, CD34, Snail, Slug, and CD105 expression among purified SSEA3+ cells revealed that expression of these markers was lacking in all SSEA3+ cells (Fig. 3E–K); however, the mesenchymal stem cell marker CD105 was expressed in 92.6% of SSEA3+ cells (Fig. 3K). We evaluated the reprogramming of SSEA3+ cells in culture, and approximately 78.2% (third generation) and 62.6% (eighth generation) of purified SSEA3+ cells remained positive for SSEA3 (Fig. 3L and M). The SSEA3− cells that were sometimes purified were rarely found to express SSEA3 (Fig. 3N).
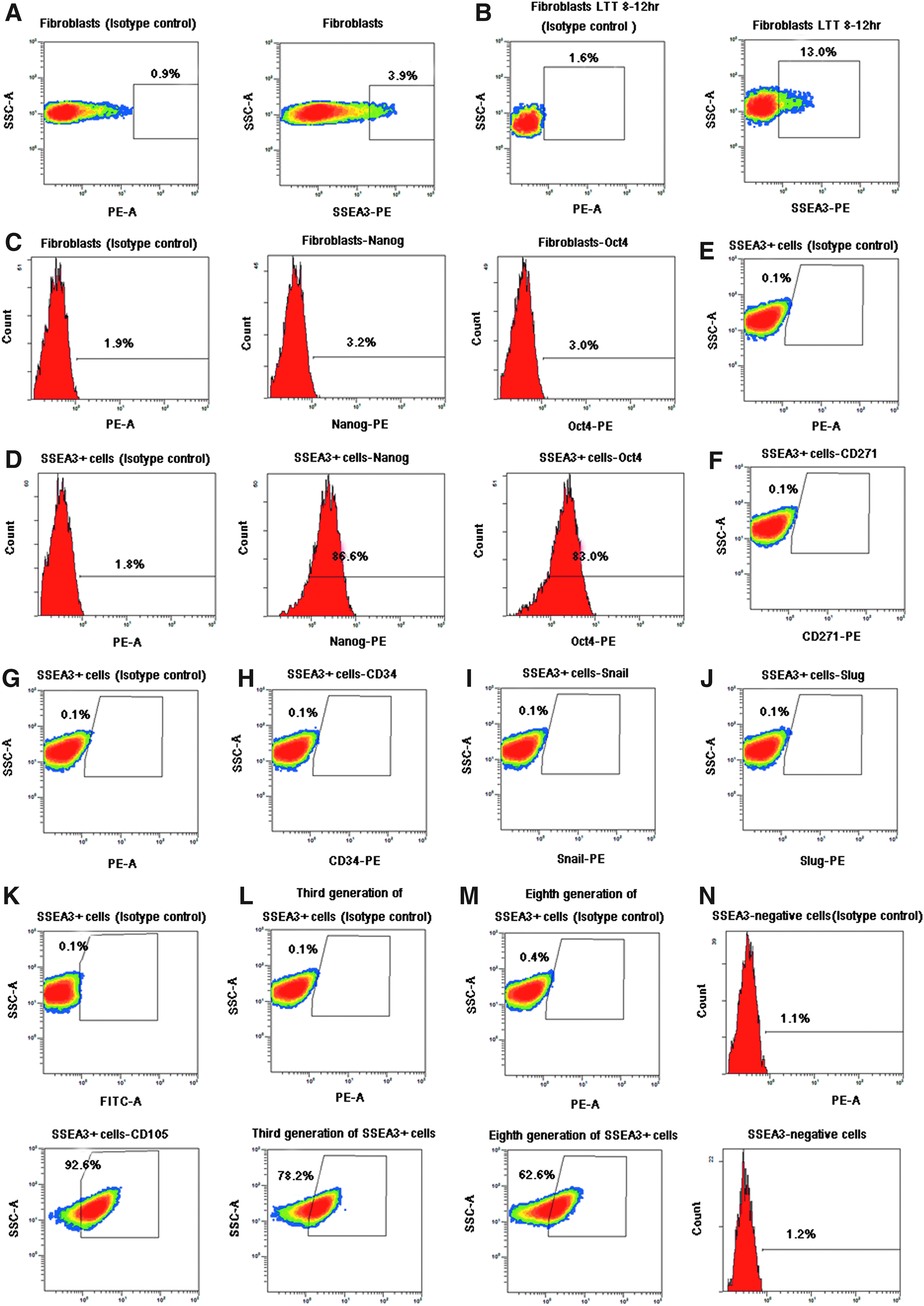
(
In vitro differentiation potency of goat SSEA3+ cells
To assess the ability of differentiation in vitro, cell clusters at P1 and P3 were transferred onto gelatin-coated dishes. Neural stem cell–like spheres appeared when the cells were induced for 7–10 days in Neurobasal medium. Immunocytochemistry revealed that these spheres were positive for the neural stem cell marker nestin (Fig. 4A, B). However, when these spheres were transferred onto gelatin-coated dishes to induce neural cells, no cells were negative for neurofilament and β-III tubulin by immunofluorescence staining.
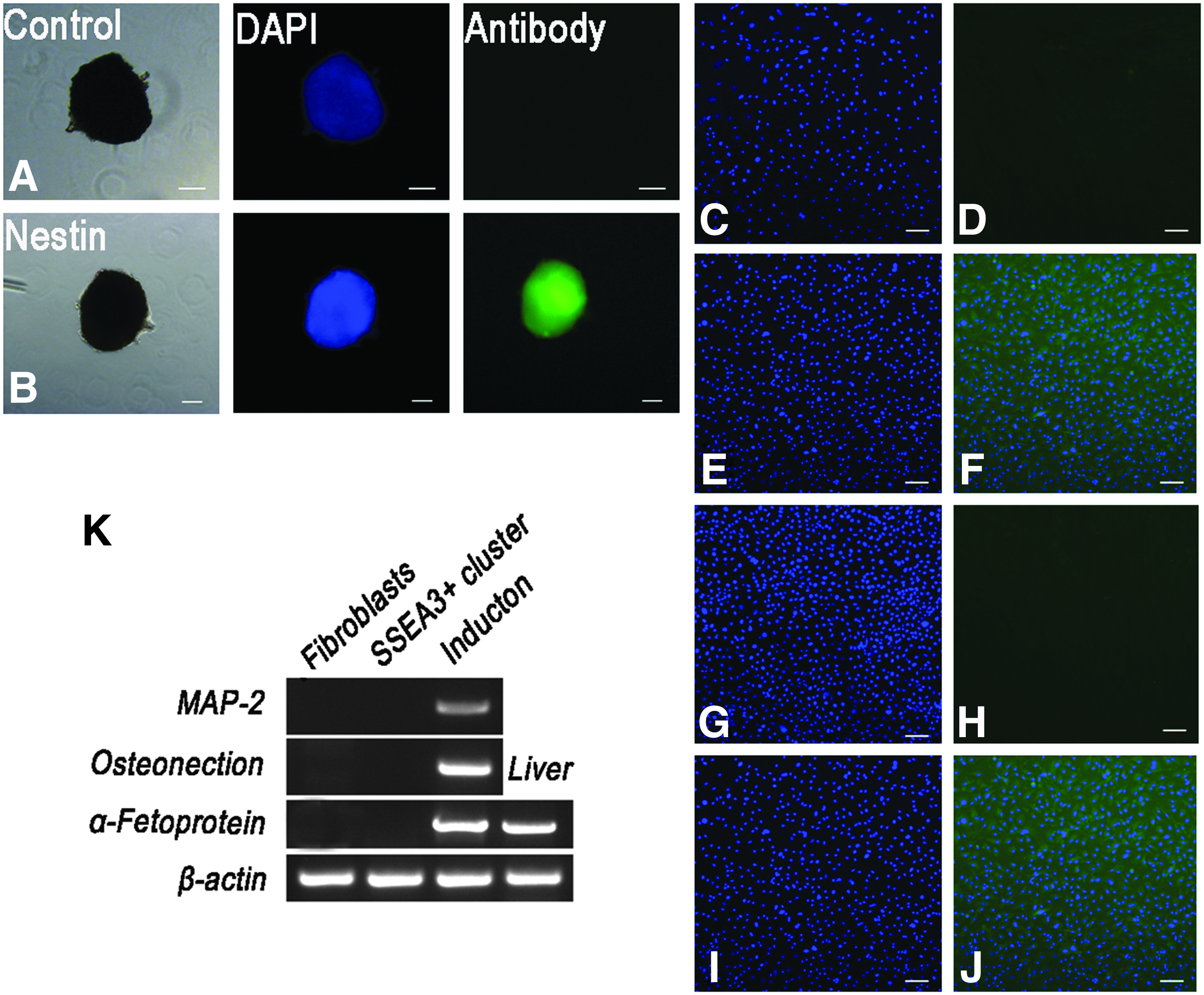
Differentiation of goat SSEA3+ cells in vitro. (
Osteoblastic induction resulted in cells positive for the mesoderm marker ALP on day 16 (Fig. 4E, F), and hepatocytic induction resulted in cells positive for the endodermal marker α-fetoprotein on day 14 (Fig. 4I and J). RT-PCR analysis also confirmed that induced cells expressed the ectodermal marker microtubule-associated protein 2 (MAP-2), the mesodermal marker osteonection, and the endodermal marker α-fetoprotein. These markers were not clearly detected in goat fibroblasts (Fig. 4K). Goat fibroblasts cultured in the same induction medium were unable to differentiate into cells from all three germ layers. On day 3, fibroblasts began to go through necrosis, and fibroblasts were no longer viable by day 10 in culture.
In vivo differentiation potential of goat SSEA3+ cells
To further evaluate the pluripotency of goat SSEA3+ cells, we injected these cells into the hind limbs of nude mice. In addition, J1 mESCs and MEFs treated with mitomycin C were used as positive and negative controls, respectively. Mice injected with MEFs did not form teratomas for up to 12 weeks (Fig. 5A). The mice administered goat SSEA3+ cells and J1 mESCs formed teratomas after 8 weeks (Fig. 5B–E). H&E staining of the teratomas showed that SSEA3+ cell–derived teratomas had a typical structure representative of all three germ layers in vivo, including stratified squamous structures (ectoderm), adipose tissue (mesoderm), and gut-like structures (endoderm) (Fig. 5F–H).
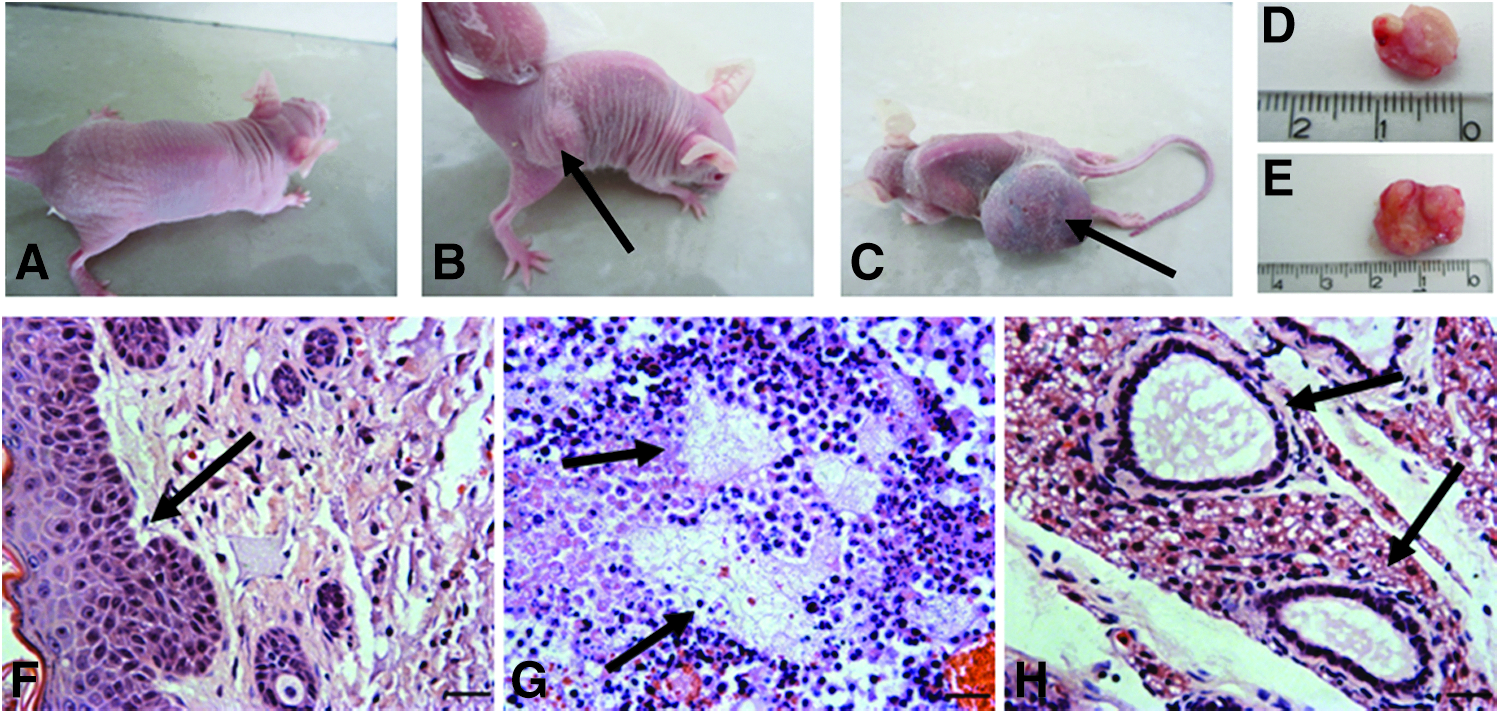
Teratoma formation. (
Discussion
Stem cells show a great potential for cell therapy and regenerative medicine, because stem cells have a capacity for self-renewal and can differentiate down multiple pathways. Recent studies have demonstrated that a population of SSEA3+ cells exists in human dermal tissues (Kuroda et al., 2010). These SSEA3+ cells form clusters in suspension cultures and are positive for pluripotency markers and AP. They are also able to differentiate into cells from all three germ layers in vitro and in vivo. It has also been shown that iPSCs can only be generated from SSEA3+ cells and not from SSEA3− cells. The efficiency of iPSCs formed (0.03%) was 30 times more efficient than for fibroblasts (approximately 0.001%) (Byrne et al., 2009; Wakao et al., 2011). These findings help to further elucidate the mechanism of iPSCs generation and increase the efficiency of reprogramming. In this study, we isolated 3–4% SSEA3+ cells from adult goat stem cells. Adult goat SSEA3+ stem cells are able to self-renew, are positive for AP and pluripotency markers (Nanog, Oct4, and Sox2), and can differentiate into endodermal, mesodermal, and ectodermal cells in vitro and in vivo.
Morphologically, the adult goat SSEA3+ cells described here are indistinguishable from general fibroblasts in adhesion culture. When the cells were transferred into suspension culture at a density of less than 5000 cells/mL, cell clusters formed with diameters of 20–150 μm. However, when cell clusters were transferred onto gelatin-coated dishes, cells expanded from these clusters grew in a manner similar to fibroblasts in adherent culture. Again, after suspension culture, cell clusters appeared from the expanded cells. Thus, we concluded that a subpopulation of SSEA3+ cells were resident among goat skin fibroblasts. We confirmed this through our flow cytometry analysis, with our data consistent with those previously published (Kuroda et al., 2010).
Our findings have also revealed that suspension cultures are a good technique for enriching skin stem cells. Chen and Pei (2012) proposed that suspension cultures might affect cell senescence or proliferation in different ways. This is critical for successful reprogramming. Thus, reprogramming in suspension culture is likely to provide insight into new possibilities for future basic and applied research.
We showed that SSEA3+ cells can tolerate stressful conditions, in particular LTT. Stressful conditions increased the numbers of stem cells; Hong et al. (2009) and Qiu et al. (2009) also found that under stressful or damaging conditions, dormant tissues stem cells became activated. Therefore, we conclude that some minimal level of stress can enrich the numbers of stem cells. In our study, LTT for 8–12 h appeared to be the optimal period for subjecting cells to stress.
Goat SSEA3+ stem cells are negative for CD34, CD271, Snail, and Slug, suggesting that these stem cells differ from those found in adult skin tissue. However, we found that the majority of SSEA3+ cells were positive for CD105, a known marker of mesenchymal stem cells (Barry et al., 1999; Dominici et al., 2006). We hypothesize that the majority of SSEA3+ stem cells belong to the CD105+ mesenchymal cell population, although their origin requires further investigation.
Although several kinds of stem cells are present in skin tissue, such as NCSCs, PCs, EPs, SKPs, and ADSCs, these stem cells only can differentiate into ectodermal and mesodermal lineage cells (Biernaskie et al., 2009; Crisan et al., 2008; Fernandes et al., 2004; Gimble et al., 2007; Middleton et al., 2005; Murga et al., 2004; Nagoshi et al., 2008). In our study, goat SSEA3+ cells differentiated into neural lineage cells and gave rise to osteoblasts and hepatocytes. This shows that the SSEA3+ stem cells were different from other adult stem cells, and perhaps they may even be unique. On the basis of the work of others (Pan et al., 2008; Pittenger et al., 1999), we suspect that SSEA3+ cells potentially contribute to such multilineage differentiation of mesenchymal stem cells. Contrary to earlier reports, tumors were observed when cells were injected into nude mice. H&E staining showed that SSEA3+ cells could differentiate into endodermal, mesodermal, and ectodermal lineage cells, thereby fully demonstrating the pluripotency of SSEA3+ cells.
Currently, studies on skin stem cell properties are hampered by a lack of suitable surface markers that can be used to identify and isolate them. It is unlikely that a single cell-surface marker could be used to separate a pure population of skin stem cells. The most important finding from our work is that adult goat stem-like cells can be efficiently isolated from naïve tissues based on SSEA3 positivity. SSEA3 is a cell-surface glycosphingolipid that plays an important role in identifying cancer cells (Schrump et al., 1988) or stem cells (Shevinsky et al., 1982). We believe that it can be effectively used as a candidate surface marker to identify goat skin stem cells.
Recent research increasingly suggests that there is some relationship between stem cells and somatic cell nuclear transplantation (SCNT). Stem cells possess higher efficiency as somatic cell nucleus donors for somatic cell cloning compared with ordinary fibroblasts (Dutta et al., 2011; Faast et al., 2006; Inoue et al., 2007; Jaenisch et al., 2002; Zhu et al., 2004). Genetic modification by homologous recombination is higher in ESCs than in somatic cells (Piedrahita 2000). To date, it has been difficult to generate definitive ESCs or equivalent iPSC lines from domestic animals. Furthermore, Wakao et al. (2011) and Byrne et al. (2009) proposed that only SSEA3+ cells can be successfully induced into iPSCs. It has been hypothesized that undifferentiated stem cells may be easier to reprogram. We aim to conduct further research and hope to generate goat iPSCs from goat SSEA3+ cells and to explore whether cloning efficiency in animals can be improved by using adult stem cells as donor nuclei for SCNT. It is possible that SSEA3+ cells will be a viable source of cells that could lead to the development of transgenic biotechnologies, which will further help elucidate the mechanisms of cellular reprogramming.
In conclusion, we isolated goat SSEA3+ stem cells from terminally differentiated goat fibroblasts cells in suspension culture. These expressed markers of pluripotent stem cells could be maintained in an undifferentiated state in vitro and were able to differentiate into cells of all three germ layers cells in vitro and in vivo. This is the first report of adult goat SSEA3+ multipotent cells in suspension culture. The detection of goat skin stem cells offers a potential resource for investigating mechanisms of cellular reprogramming.
Footnotes
Acknowledgments
The authors are grateful to Dr. Bin Wu at the Arizona Center for Reproductive Endocrinology and Infertility for critically reading and revising our manuscript. We also thank Mr. Jintao Hu for technical assistance with flow cytometry. This work was supported by the National High Technology Research and Development Program of China (863 Program) (No. 2011AA100303).
Author Disclosure Statement
The authors declare no conflicts of interest.
Animal welfare and experimental procedures were carried out in accordance with the Guide for the Care and Use of Laboratory Animals (Ministry of Science and Technology of China, 2006) and were approved by the animal ethics committee of Northwest A & F University.