
Abstract
Abstract
In this study, we tested the effects of valproic acid (VPA), a known histone deacetylase inhibitor (HDACi), on the growth characteristics, apoptosis, and cell cycle stages distribution of donor cells, as well as cloning efficiency, embryo development, and histone methylation. Our results showed that treatment of donor cells with VPA (2.5 mM, 5.0 mM, 7.5 mM, or 10 mM) for 24 h resulted in altered cell proliferation, extent of apoptosis and necrosis, and cell cycle stage distribution, whereas no changes in cell viability and chromosomal complements were observed. Measurement of relative gene expression using real-time PCR of a few developmentally important genes in treated donor cells showed decreased expression of HDAC1 and increased expression of BAX (p<0.05). No change in relative expression of HDAC2 and Bcl2 was noticed. Treatment of donor cells with VPA for 24 h before electrofusion significantly (p<0.05) increased the blastocyst formation rate of somatic cell nuclear transfer (SCNT) embryos compared to the control embryos. Terminal deoxynucleotidyl transferase dUTP nick end labeling (TUNEL)-positive nuclei in SCNT blastocysts derived from VPA-treated donor cells were significantly decreased compared to the control blastocysts (p<0.05). Immunolocalization studies revealed that the levels of histone H3 at lysine 9 (H3K9me3) were lower in VPA-treated donor cells derived cloned blastocysts than nontreated cloned embryos, and was at the level of in vitro fertilization (IVF) counterparts, although no effects of treatments were found in donor cells. Our study demonstrates that the use of VPA in SCNT has been beneficial for efficient reprogramming of donor cells. Its effect on histone methylation in cloned embryos correlates with their developmental potential and may be a useful epigenetic marker to predict the efficiency of SCNT.
Introduction
Recently, several histone deacetylase inhibitors (HDACi) such as the trichostatin A (TSA) (Iager et al., 2008; Li et al., 2008; Wang et al., 2011), Scriptaid (Zhao et al., 2009; Van Thuan et al., 2009; Zhao et al., 2010), sodium butyrate (Das et al., 2010; Shi et al., 2003; Yang, F.K., et al., 2007), suberoylanilide hydroxamic acid (SAHA) (Ono et al., 2010), m-carboxycinnamic acid bishydroxamide (CBHA) (Dai et al., 2010), and Oxamflatin (Su et al., 2011) have been used to improve the developmental competence of SCNT embryos. Results have indicated that HDACi significantly improve the in vitro and full-term development of SCNT embryos. Few reports suggested that the commonly used HDACi TSA has an adverse effect on development of cloned embryos (Meng et al., 2009; Wu et al., 2008), thus finding a new potent alternative for TSA is of paramount importance in SCNT research. We have explored whether VPA can be used as an effective reprogramming modulator.
VPA, a kind of short-chain fatty acid with the chemical name 2-sodium valproate, is widely used as an anticonvulsant drug and it has been categorized as a specific HDACi. Recently, VPA was used as a reprogramming drug for mouse embryonic fibroblasts (MEFs) and human fibroblasts (Huangfu et al., 2008a, b). VPA usage also resulted in improvement of cloning efficiency in mouse, miniature pig, and cattle (Costa-Borges et al., 2010; Miyoshi et al., 2010; Xu et al., 2012), thereby suggesting that VPA treatment can be useful for the improvement of reprogramming events in the cloned embryos.
However, the mechanisms of action of VPA in modulating reprogramming events have not been investigated. Therefore, we have investigated the effects of VPA on nuclear reprogramming of somatic cells and elucidated the possible mechanism of how VPA treatment improves the cloning efficiency. In support of this hypothesis, we have explored the effects of VPA on the growth pattern of donor cells, including their proliferation, viability, chromosomal complement, induction of cell apoptosis, cell cycle stage distribution, expression levels of apoptosis, and development-related genes and global methylation levels of histone H3 at lysine 9 (H3K9me3). Furthermore, we have analyzed the effect of donor cell VPA treatment on SCNT embryo production rate, embryo health quality, and global methylation levels of H3K9me3.
Materials and Methods
Chemicals were obtained from the Sigma Chemical Company (St. Louis, MO, USA), media from GIBCO (Grand Island Biological Company, Grand Island, NY, USA), and disposable plastic ware from Nunc (Roskilde, Denmark), unless otherwise stated. In vitro culture of oocytes was done at 38.5°C with 20% CO2 in air, whereas embryos were cultured at 38.5°C with 5% CO2, 5% O2. The experimental study plan of the present study is shown in Figure 1.

Experimental design of the present study. PAR, IVF, and HMC represent parthenogenetic, in vitro-fertilized, and handmade cloned embryos.
Cell culture and VPA treatments
Bovine ear skin fibroblasts were derived from an adult male bull and cryopreserved in Dulbecco's modified Eagle medium (DMEM; Gibco, Grand Island, NY, USA) supplemented with 10% fetal bovine serum (FBS), and 10% dimethyl sulfoxide (DMSO) in cryovials. During experimental study, cells were thawed, cultured, and maintained in DMEM supplemented with 10% FBS and 1% (vol/vol) penicillin-streptomycin (Gibco BRL, Burlington, ON, Canada) at 37°C in 5% CO2 in air and were used for VPA treatments and SCNT. During each experiment, donor cells were seeded at a concentration of 1.5×105 cells per well on a six-well culture plate and grown to 70–80% confluence. After 24 h of culture, the cells were incubated in the culture medium containing a selected dose of VPA (2.5 mM, 5.0 mM, 7.5 mM, 10 mM; Sigma) for additional 24 h. After that they were subjected to various experiments as shown in Figure 1. All experiments were conducted on passages 5–10 number to minimize the difference in results due to aging caused by subpassage.
Morphological changes, proliferation, and viability of VPA-treated donor cells
Cells were treated with different concentrations of VPA as mentioned above, and nontreated cells were considered as controls. Morphological changes were observed by taking images on an Olympus inverted microscope at 20× magnification. Population doubling times were calculated by seeding the same concentration of cells/well for all groups and harvesting after 72 h of VPA treatments and were determined by freely available doubling time software (www.doubling-time.com). The proliferative activities of VPA-treated donor cells were measured by immunohistochemical staining for the Ki-67 proliferation marker using mouse monoclonal antibodies (Abcam). The labeling index of Ki-67 was expressed as the percentage of positive cells after counting ≈1000 cells in five to six fields selected randomly in each drug concentration. The Trypan Blue dye exclusion test was used to determine the number of viable cells. This test is based on the principal that live cells possess an intact cellular membrane that can actively exclude dyes, whereas dead cells do not have such integrity and so are stained in a blue color. Experimental group cells that excluded Trypan Blue after incubation with phosphate-buffered saline (PBS) containing 0.04% Trypan Blue were considered viable, and the viable cell percentage was calculated for each group.
Apoptosis and necrosis detection in VPA-treated cells
To evaluate the effect of VPA treatments on the extent of apoptosis and necrosis, cells were analyzed using annexin V and propidium iodide (PI) staining kits (Roche Diagnostics, Mississauga, ON, Canada), as described by the manufacturer's protocol with slight modifications. Briefly, treated and nontreated fibroblast cells were harvested by trypsinization and were incubated in the apoptosis and necrosis staining solution containing (10 μg/mL H-33342, 1 μL/50 μL PI, and 1 μL/50 μL annexin V in the staining buffer) for about 15–20 min. Subsequently, the cells were washed in Dulbecco's phosphate-buffered saline (DPBS), mounted on a clean glass slide in a drop of Vectashield (Vector Laboratories, Burlingame, CA, USA), and flattened slowly with a coverslip. The images were taken with a Leitz fluorescence microscope (Zeiss/Leica LEO 912 AB, ON, Canada) using a blue, red, or green filter. Cells staining red were considered necrotic, whereas green cells were considered apoptotic. For exclusion of a false signal from both red and green filters, nuclear staining with Hoechst 33342 was considered at time of calculation. A minimum of five different image fields were examined for each experiment, and three independent experiments were performed. The percentages of apoptotic and necrotic cells were calculated for each group as follows: Apoptotic cell %=(number of annexin-positive cells/total number of cells)×100. Necrotic cell %=(number of PI-positive cells/total number of cells)×100.
Chromosomal analysis of VPA-treated cells
After 24 h of VPA treatment, the cells were cultured in culture medium containing 0.1 mg/mL colcemid for 4 h. After trypsinization (0.25% trypsin–EDTA), recovered cells were suspended and treated with a hypotonic solution of 0.56% KCl for 30 min at room temperature. Fixative solution (methanol:acetic acid, 3:1) was added to the cells and centrifuged at 700×g for 5 min. The fixative process was repeated twice. The cell pellet was suspended in 1 mL of fixative solution and was dropped onto the slide to produce metaphase spreads. The slides were immersed in a 0.5% barium hydroxide solution for 3 min and incubated in 2× SSC (30 mM NaCl and 30 mM trisodium citrate) for 2 h at 50°C. The samples were then stained with 0.1% Giemsa (Fluka, Gallen, Switzerland) for 30 min, washed three times with distilled water, and dried in air. The metaphase chromosomes were observed, and images were taken under oil emulsion using a Leitz microscope (Zeiss/Leica LEO 912 AB, ON, Canada). Chromosome numbers were counted manually.
Cell cycle stage distribution of VPA-treated cells
To evaluate the effect of VPA treatments on the distribution of cell cycle stages, DNA content was measured by flow cytometric analysis using PI staining, as described previously (Darzynkiewicz and Huang 2004) with slight modifications, and the relative proportions of cells in the G0/G1 (2C DNA content), S (2C–4C), and G2/M (4C) stages were calculated. Briefly, treated and nontreated fibroblast cells were harvested by trypsinization and fixed in chilled 70% ethanol overnight. The cells were then incubated in DNA staining solution containing (0.2 mg/mL RNase, 20 μg/mL PI, and 0.1% Triton X-100) for 10 min. Subsequently, the cells were transferred in DPBS, and flow cytometric analysis of the cell cycle was performed using a 488-nm laser line for excitation in a BD FACS Calibur (Becton-Dickinson, Rutherford, NJ, USA). For each cell treatment, 10,000 cells were analyzed, and the proportion of cells in G0/G1, S, and G2/M stage was estimated using the Flowjo Cycle Analysis program (Tree Star Inc., Ashland, OR) with the same algorithm in all samples. At least three independent flow cytometry experiments were performed for each dose of VPA.
Gene expression analysis in VPA-treated cells
Total RNA of the nontreated controls and the VPA-treated cells was extracted using the conventional TRIzol (Invitrogen, Burlington, Ontario, Canada) procedure with an extra step of DNase I treatment for the removal of DNA contamination. The concentration of extracted total RNA was determined by measuring the absorbance at 260 nm in a spectrophotometer. Reverse transcription (RT) reactions were performed on the RNA extracted from groups using 500 ng of oligo(dT) and Superscript II (Invitrogen) reverse transcriptase. The oligo(dT) was first added to the RNA samples and allowed to anneal by denaturing the secondary structures at 70°C for 2 min. A mixture of 4 μL of RT buffer, 1 μL of 0.1 M dithiothreitol, 1 μL of 10 mM deoxyribonucleotide triphosphates (dNTPs) mix, 0.5 μL of RNasin (40 U/μL) (Promega, Madison, WI, USA), and 1 μL of Superscript II (200 U/μL) (Invitrogen) was added and the reaction was incubated at 42°C for 1 h, followed by a denaturing step at 70°C for 30 min. The real-time RT-PCR was carried out using the LightCycler 3.0 and the LightCycler FastStart DNA Master SYBR Green I (Roche), according to the manufacturer's protocols. Each reaction mix contained 1 μL of the cDNA reaction, 1 μL of the FastStart DNA Master SYBR Green I reaction mix, 3 mM MgCl2, and 1 μL of each the forward and reverse primers (GAPDH, forward, 5′-TTCCT GGTACGACAATGAATTTG-3′, reverse, 5′-GGAGATGGGG CAGGACTC-3′; BAX, forward, 5′-CCACACACACACACA TTCATTC-3′, reverse, 5′-TCATACAACACGGGCTTCCT-3′; Bcl2, forward, 5′-TGTGTGTGGAGAGCGTCAAC-3′, reverse, 5′-CAGCCAGGAGAAATCAAACAG-3′, and HDAC1, HDAC2; Beyhan et al., 2007), adjusted to a total volume of 10 μL using H2O. Histone-modulating genes (HDAC1 and HDAC2), apoptosis genes (Bcl2 and BAX), and internal control genes (GAPDH) were analyzed for the pattern of the transcripts by quantitative real-time RT-PCR as described previously (Favetta et al., 2007).
Production of handmade cloned, in vitro–fertilized, and parthenogenetic embryos
Cattle (Bos taurus) ovaries were collected from a government-inspected abattoir (Better Beef Ltd., Guelph, ON, Canada), and oocytes were aspirated using a vacuum pump and subjected to in vitro maturation, as previously described by (Favetta et al., 2007; Mastromonaco et al., 2007). After maturation, cumulus–oocyte complexes (COCs) were subjected to the process of handmade cloning (HMC) as described previously (Selokar et al., 2012), with slight modifications. Briefly, in vitro–matured cumulus oocyte complexes with expanded cumulus were stripped off their cumulus investment and zona pellucida using hyaluronidase (0.5 mg/mL in sperm tap) and Pronase (1.0 mg/mL in DPBS), respectively. Oocytes with completely digested zona pellucida were transferred to T20 [tissue culture medium-199 (TCM-199) containing 20% FBS] and incubated at 38.5°C for 15–20 min so that a prominent protrusion cone was easily visible. Protrusion cone–guided bisection was performed using a cutting blade (Labco, Ambala, India) in 3 mL of T20 containing 2.5 μg/mL cytochalasin B, followed by Hoechst 33342 staining confirmation of removal of the metaphase II (MII) plate.
The larger demicytoplasts without a protrusion cone were transferred to T20 and incubated at 38.5°C to enable them to regain a spherical shape. The enucleated demicytoplasts were immersed in phytohemagglutinin (0.5 mg/mL in T2) for 3–4 sec and then transferred to T2 containing donor cells at a low cell density. Each demicytoplast was then allowed to attach to a single, rounded, medium sized cell by gently rolling the demicytoplast over it. The couplets (demicytoplast–donor cell pairs) were transferred to fusion medium (0.3 M
The reconstructed oocytes were activated by incubating in T20 containing 4 μM Ca ionophore A23187 for 5 min at 38.5°C. After washing three times with T20, the reconstructed oocytes of were transferred individually to 5-μL droplets of T20 containing 2 mM of 6-dimethylamino purine, covered with mineral oil, and incubated for 4 h in a CO2 incubator at 38.5°C. The reconstructed, activated embryos were washed twice and then cultured in 500 μL of synthetic oviductal fluid (SOF) (Chemicon-Millipore, Billerica, MA, USA) supplemented with 200 μL of nonessential amino acids, 100 μL of essential amino acids (Invitrogen), 5 μL of gentamicin (Invitrogen), 560 μL of fatty acid–free bovine serum albumin (FAF BSA; 15%) in SOF and 200 μL of bovine steer serum. The four-well dishes (15–20 embryos per well) were covered with mineral oil and kept undisturbed under 38.5°C in a CO2 incubator at 5% CO2, 5% O2, and 90% N2 in air for 8 days. Parthenogenetic activation and culture of zona-free oocytes were carried out just as for NT oocytes, and in vitro fertilization was as described previously (Mastromonaco et al., 2007)
Assessment of fusion efficiency, embryo development, determination of blastocyst cell number, and apoptosis
Fusion efficiency was checked after 30 min of reconstruction; unfused/lysed embryos were discarded, and the remaining intact fused embryos underwent a process of activation. Rates of embryo development (cleavage and blastocysts) were recorded on day 8 of IVC and percent development of each stage was determined. Cytogenetic analysis of day-8 cloned and parthenogetic blastocysts was examined and efficiency of the cloned embryos production protocol was confirmed as described by King et al. (2004). For examining the health of the embryos, the total cell number and apoptosis of day-8 blastocysts were determined by terminal deoxynucleotidyl transferase dUTP nick end labeling (TUNEL) staining as described previously (Brison and Schultz, 1997). Briefly, cloned blastocysts from both control and treatment groups, along with parthenogenetic and IVF blastocysts, were washed separately three times in PBS containing 0.3% polyvinyl alcohol (PVA) in four-well dishes and fixed in 4% paraformaldehyde for 1 h at room temperature and followed by three washing steps. The blastocysts were kept at 4°C until starting the staining procedure. Prior to staining, the blastocysts were washed three times in PBS–0.3% PVA to remove any traces of paraformaldehyde. Blastocysts were then permeabilized by incubation in 0.5% Triton X-100 for 1 h and further incubated with fluorescein isothiocyanate (FITC)-conjugated 2′-deoxyuridine, 5′-triphosphate (dUTP) and terminal deoxynucleotidyl transferase (TdT) enzyme for 1 h at 37°C in the dark, treated with 50 μg/mL RNase at room temperature for 1 h, and counterstained with 10 μg/mL propidium iodide (PI) for 30 min at 37°C. Positive and negative controls were processed along with control and treatment groups. For the positive controls, blastocysts were incubated in DNase solution (100 U/mL) for 20 min at 37°C before enzyme solution incubation. Stained blastocysts were then washed in DPBS (calcium and magnesium free) and mounted on a glass microscope slide in a 3-μL drop of Vectashield Anti-Fading solution (Vector Laboratories, Burlingame, CA, USA) and flattened with a cover slip. Images were captured with both red and green filters for nuclei and site of apoptosis, respectively, and merged images were generated from red and green filters showing a yellow body on the exact site of red nuclei considered as apoptotic cells. Cell counting was performed from digital images obtained on an upright Leitz fluorescence microscope. Total apoptotic indices were calculated for each embryo as follows: Apoptotic index of blastocyst=(number of TUNEL-positive nuclei/total number of nuclei in blastocyst)×100.
Global H3K9me3 analysis in donor cells and blastocysts
Global H3K9me3 analysis in donor cells and blastocysts was done using the immunofluorescence technique described by Madan et al. (2005) with slight modifications. Briefly VPA-treated donor cells and generated cloned blastocysts were fixed in 4% paraformaldehyde (PFA) in PBS for 1 h at room temperature and then washed three times in PBS–0.3% PVA before permeabilization in 0.5% Triton X-100 in PBS for 1 h at room temperature. Cells and blastocysts were blocked overnight at 4°C in 5% BSA–0.3%VPA and 0.1% Triton X-100 in PBS before incubation with primary antibody. Cells and blastocysts were then incubated for 1 h with the primary antibody, mouse anti-H3K9me3 (Abcam) diluted 1:200, in PBS with 5% BSA at 37°C. After washing with PBS, cells and blastocysts incubated with a FITC-conjugated donkey anti-rabbit secondary antibody (Sigma-Aldrich) diluted 1:500 in PBS with 5% BSA. After thorough washing, the nuclei were counterstained with 4′,6-diamidino-2-phenylindole (DAPI; 1 μg/mL) and rinsed in PBS–0.3% PVA. Finally cover slip–grown cells and embryos were mounted on slides in Vectashield fluorescence mounting medium. The slides were observed under a Leitz fluorescence microscope (Zeiss/Leica LEO 912 AB, ON, Canada), and images were captured keeping the same optical conditions. Openlab Leica image processing software (Improvision, Lexington, MA, USA) was used for image acquisition and quantitative measurements of the mean pixel intensity emitted by each individual nucleus. At least 1000 nuclei of somatic cells and 10 blastocysts were analyzed for each treatment.
Statistical analysis
The percentage data were analyzed using SYSTAT 7.0 (SPSS Inc., Chicago, IL, USA) after arcsine transformation. Differences between means were analyzed by one-way analysis of variance (ANOVA) followed by the Fisher least significant difference (LSD) test for percentage data of cell cycle stages, viability, proliferation rate, apoptosis, gene expression, embryonic developmental stages, and signal intensity. Differences were considered to be significant at p<0.05.
Results
Effect of VPA treatment on donor cells growth pattern and health status
Prior to HMC, we tested the effect of VPA treatment on growth characteristics of donor cells. We found that VPA-treated cells were enlarged in size, flattened, and elongated in outlines with a significant increase in population doubling time in a dose-dependent manner (p<0.05), which was also supported by significant decrease in the Ki-67 proliferation index (Fig. 2). Primary consideration during our HMC procedure was the proportion of healthy cells in each treatment dose and, therefore, we conducted two tests—viability assessment using Trypan Blue dye exclusion and a apoptosis/necrosis test using an annexin V/PI assay. We found that VPA treatment induced apoptosis at 5 mM and higher doses with increasing trend (p<0.05). No significant effect on proportion of necrotic cells (PI positive) and late apoptotic cells (annexin/PI positive) was noticed. Although VPA induced apoptosis, no significant effect on cell viability was noticed as indicated by Trypan Blue dye exclusion method (Fig. 2) and chromosomal complement (Fig. S1). (Supplementary Data are available at www.liebertpub.com/cell/.)

Effects of VPA treatment on the growth pattern of donor cells. (
Analysis of cells cycle stage distribution in VPA-treated donor cells
To evaluate the effect of VPA treatment on the donor cell cycle stage status, DNA content was measured by flow cytometric analysis, and the relative percentage of the proportions of cells in the G0/G1 (2C DNA content), S (2C–4C), and G2/M (4C) stages were calculated. A 2.5 mM dose of VPA had little effect on cell cycle–stage distribution status, whereas VPA doses of 5.0 mM, 7.5 mM, and 10 mM significantly altered cell cycle stage distribution (p<0.05) and induced cell at G0/G1 stage in a dose-dependent manner, with concomitant decrease in the proportion of these cells in the S stage of the cell cycle (Fig. 3).

Cell cycle stage distribution of VPA-treated donor cells. Arrow indicates population of S-phase cells. SSC, FSC, and FL2 represent side scatters, forward scatters, and filter position. Different superscripts differ significantly (p<0.05).
Analysis of relative expression of histone modulating and apoptosis genes in VPA treatment of donor cells
Relative expression levels of two histone-modulating and two apoptosis-related genes were analyzed in VPA-treated donor cells using quantitative real-time PCR. The expression level of HDAC1 was significantly lower (p<0.05) in all treated donor cells groups as compared to the nontreated cells, although no change was evident in the expression pattern of HDAC2 gene (Fig. 4). In apoptotic genes, the expression level of Bax was higher in 5 mM, 7.5 mM, and 10 mM VPA-treated cells than that for 2.5 mM VPA-treated and nontreated cells (p<0.05). However, the expression level of Bcl2, was not affected by any dose of VPA treatment when compared to the control (p<0.05).

Relative expression levels of chromatin remodeling- (
In vitro developmental competence and health of HMC embryos derived from VPA-treated donor cells
We tested the efficiency of our nuclear transfer protocol using the fluorescent in situ hybridization (FISH) method, which indicated that our method of nuclear transfer produced 100% cloned embryos (Fig. S2). To assess whether altering the epigenetic status of donor cells could benefit in vitro development of HMC bovine embryos, we performed SCNT of treated somatic cells used as donor. We found that in all HMC group embryos fusion and cleavage rates were not significantly affected. However, significant improvement (p<0.05) in blastocyst production rates with donor cells treated with 2.5 mM VPA among all tested groups was observed (Table 1). For testing the effect of VPA on the health of embryos, TUNEL assay (Fig. 5) indicated that blastocysts generated from 2.5 mM VPA-treated donor cells had lower apoptotic index, although no significant effect on total cell number among groups was noticed (Table 1). Even though the apoptosis rate was lowered by VPA treatments, this lowering was not at the same extent as that of in vitro–fertilized counterparts.

TUNEL assay for determination of apoptosis extent and total cell number in VPA-treated donor cell-derived cloned embryos. FITC-conjugated dUTP-labeled nuclei were considered as apoptotic, and PI-stained nuclei were used for determining total cell number. In negative controls, only labeled solution was used, whereas for positive controls DNase treatment was done before incubating with a mixture of FITC-conjugated dUTP and terminal transferase enzyme. Staining in the merged projection of the positive controls and test samples indicates DNA fragmentation and apoptosis.
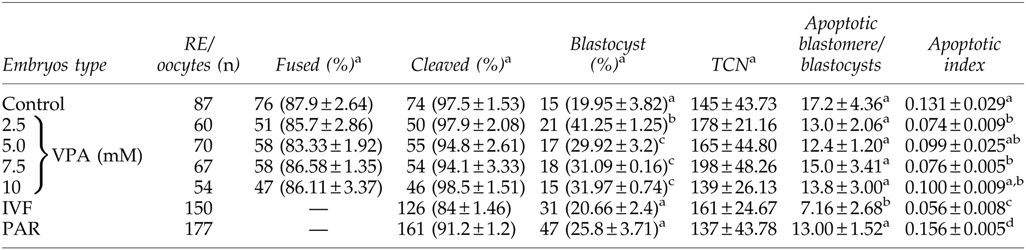
Data from 5–7 trails. Values are mean±standard error of the mean (SEM.)
Values with different superscripts within the same column are significantly different (p<0.05).
Parthenogenetic embryos were considered as internal controls for activation protocol and oocyte quality.
RE, reconstructed embryos; TCN, total cell number in blastocyst; VPA, valproic acid; IVF, in vitro–fertilized; PAR, parthenogenetic.
Histone methylation levels H3K9 in donor cells and cloned blastocysts
To find the way in which VPA treatment improved the developmental competence of HMC embryos, the methylation levels of the H3K9 epigenetic marker were studied in donor cells and blastocyst-stage embryos. As shown in Figure 6, VPA treatments do not affect H3K9me3 levels in donor cells of all treatment groups (p>0.05). H3K9me3 levels in HMC blastocysts were significantly lower in all of the selected doses, which were similar to the levels of IVF-produced counterparts (p<0.05). However, parthenogenetic blastocysts had high levels of H3K9me3 among all groups (Fig. 7). IVF embryos with normal morphology were considered as normally reprogrammed embryos, whereas parthenogenetic embryos were considered as controls for the oocyte quality and activation protocols.

Effect of VPA on cell morphology and global histone methylation at the ninth Lysine residue of histone 3 nuclear protein (H3K9me3) in donor cells. Cells were treated with indicated concentrations of VPA and analyzed for morphological changes and H3K9me3 by semi-quantitative immunofluorescence. Mean pixel intensity of H3K9me3 was divided by nuclear intensity emitted from the nuclei of cells treated with each concentration of VPA. The intensity ratios for cells and blastocysts were converted into percentages for comparison.

Global H3K9me3 levels in the VPA-treated donor cells that were used to produce cloned, IVF, and parthenogenetic (PAR) blastocysts. IVF blastocysts were used because the reprogramming process is considered close to normal in such embryos, whereas parthenogetic blastocysts were used as internal controls for oocyte quality and activation protocol. At least 1000 nuclei of somatic cells and 10 blastocysts were analyzed for each treatment.
Discussion
Global epigenetic reprogramming has been reported as a major event that occurs following SCNT, regulating the success or failure of the cloning process. Previous studies demonstrated that differentiated somatic cell nuclei are in a highly methylated state and appear to be incompletely reprogrammed in cloned embryos during preimplantation development (Enright et al., 2003). Hence, modifying the epigenetic status of donor cells may be considered as one of the most important factors for improving the cloning success in farm animals. Considering this, we examined whether HDACi (VPA) treatments can improve reprogramming of somatic cells and have an effect on SCNT embryos development in vitro. We found that VPA treatment of donor cells alters cell morphology, proliferation, cell cycle stage distribution, and gene expression patterns. Use of VPA-treated donor cells in SCNT significantly improved the blastocyst production rate with minimum apoptosis and significantly modified the epigenetic status of SCNT embryos by modifying their methylation pattern (H3K9me3), almost similar to that of the IVF counterparts. Thus, we concluded that VPA treatment of donor cells is beneficial for improvement in developmental competence and enhanced nuclear reprogramming of the donor cell genome.
Genomic methylation status of cloned embryos closely resembles that of donor cells and is generally hypermethylated as compared to in vitro– or in vivo–fertilized counterparts (Bourc'his et al., 2001; Dean et al., 2001; Kang et al., 2001). Other studies have provided evidence that this may be due to incomplete reprogramming of donor nuclei (Dean et al., 2001; Morgan et al., 2005; Rybouchkin et al., 2006; Zhang et al. 2009). During the process of nuclear reprogramming, there is erasure of some of the epigenetic programming of the donor genome, which leads to the establishment of new embryonic epigenetic patterns involving DNA demethylation and methylation events, as well as other chromatin alterations (Morgan et al., 2005). Many epigenetic modifiers have been used to improve process of nuclear reprogramming (Dai et al., 2010; Zhang et al., 2007; Zhao et al., 2009). In bovines, use of 5-aza-2-deoxycytidine (AzaC) and TSA in the SCNT process improves the reprogramming efficiency (Enright et al., 2003).
This finding leads to the use of chromatin modifiers for SCNT, and that has increased the success rates and also helped in achieving significant improvements in the reprogramming process (Enright et al., 2005; Iager et al., 2008; Su et al., 2011; Wang et al., 2011). Use of HDACi in cloned embryos of different species such as mouse (Kishigami et al., 2006), rabbit (Shi et al., 2008), pigs (Cervera et al., 2009; Martinez-Diaz et al., 2010; Zhao et al., 2009; Zhao et al., 2010), and cattle (Ding et al., 2008; Iager et al., 2008; Su et al., 2011) has produced consistently promising results. VPA, a kind of short-chain fatty acid, was used for successful reprogramming of mouse and human fibroblasts cells into induced pluripotent stem cells (iPSCs) using three and two transcription factors, respectively (Huangfu et al., 2008a, b). Also its use in mouse and miniature pig resulted in the improvement of overall cloning efficiency (Costa-Borges et al., 2010; Miyoshi et al., 2010). In the present study, we hypothesized whether treatment of donor cells with VPA prior to NT results in better reprogramming and cloning efficiency and further elucidated the molecular mechanism of this reprogramming process.
In general, HDACi promote global chromatin acetylation, which leads to excessive gene transcription. This is usually translated as an increase in protein production that would further induce cellular differentiation, modifying cell growth characteristics (Goodsell et al., 2003). HDACi have also been reported to have antiproliferative and apoptotic effects, which are beneficial in cancer therapies. Mutation of the HDAC1 gene in mice caused reduced cell proliferation, leading to the embryonic death (Lagger et al., 2002). These effects were associated with increased gene expression of cell cycle inhibitors (Lagger et al., 2002). Similar antiproliferative effects were seen in embryonic stem cells (Saunders et al., 1999; Lee et al., 2004). In agreement with the above studies, we also found that use of VPA in adult bovine fibroblasts significantly altered cell morphology by inducing cellular enlargement and flattening the population doubling time (Fig. 2).
Ki-67 is a nuclear antigen associated with cellular proliferation and ribosomal RNA synthesis, and is detected exclusively within the cell nucleus, whereas in dividing cells protein is relocated to the surface of the chromosomes (Bullwinkel et al., 2006). Ki-67 antigen is present during all active phases of the cell cycle (G1, S, G2, M), but is absent from (G0) resting stage (Bullwinkel et al., 2006; Rahmanzadeh et al., 2007). Our results showed that VPA inhibits proliferation (Fig. 2), and this could be due to inhibition of ribosomal RNA synthesis and cell cycle arrest. However, no adverse effect of VPA was found on cell viability and chromosomal ploidy (Fig. 2, Fig. S1). The mechanisms underlying the effect of VPA on cell growth include both apoptosis induction and cell cycle arrest. The minimum dose of VPA responsible for the induction of apoptosis and cell cycle arrest was found to be 5.0 mM (Figs. 2 and 3). VPA significantly reduced the expression of HDAC1 in all selective doses, while increased the expression of BAX gene was found to be 5.0 mM onward (Fig. 4). There was no change in the expression pattern of the HDAC2 and Bcl2 genes. Similar effects have been seen with the use of other HDACi, such as sodium butyrate, TSA, and MS-275, in various cancer cell lines (Greenberg et al., 2001; Lucas et al., 2004; Rosato et al., 2003).
TSA is a HDACi commonly used as a reprogramming modulator in SCNT research. It has negative effects on embryo development, thus VPA might be an alternate candidate for epigenetic reprogramming of nuclear donor cells. VPA has been used to promote reprogramming of somatic cells into iPSCs by modification of chromatin remodeling (Huangfu et al., 2008a, b). It has been postulated that the process of epigenetic reprogramming in SCNT and generation of iPSCs involves chromatin modification and may share some common mechanisms (Colman and Dreesen, 2009; Kastenberg and Odorico, 2008). Chromosomes are known to retain high acetylation levels or low methylation levels and less condensed chromatin known as euchromatin when the cell stage is in the embryonic condition (Feldman et al., 2006). Most transcribed regions of DNA are found in the euchromatin. Therefore, if HDACi could change the configuration of donor cell chromosomes from heterochromatin to euchromatin, reprogramming factors may access the donor cell chromosomes easily in SCNT embryos as well as in iPSCs.
Considering the use of HDACi in SCNT production, VPA treatment of embryos after activation dramatically improved not only their blastocyst formation rate in vitro but also the production efficiency of viable cloned offsprings in mice (Costa-Borges et al., 2010). Our results also observed that developmental competence of cloned embryos was improved after the use of VPA-treated donor cells. We also observed that higher doses had a negative effect on embryo production; still, their developmental competence was higher than that of the untreated group (Table 1). This may reflect a negative effect of HDACi treatment with high concentrations, as previously described for TSA treatment (Tsuji et al., 2009). Moreover, altered cellular morphology at higher concentrations does not affect fusion efficiency. Apoptosis is natural process to eliminate cells with nuclear or chromosomal abnormalities and plays an important role in embryo development (Hardy, 1997). Apoptosis has been observed in bovine embryos after the eight-cell stage (Fahrudin et al., 2002; Matwee et al., 2000). A positive correction has been seen between incidence of apoptosis and reprogramming of donor cells in SCNT embryos (Cui et al., 2011).
In the present study, VPA SCNT embryos showed a lower apoptotic index than that of the untreated SCNT embryos, which was higher than that of IVF (Table 1). This observation is similar to that of reported previously for TSA (Cui et al., 2011). Thus, we demonstrate that blastocysts generated from VPA-treated donor cells had a lesser number of apoptotic nuclei, and this may be one of the signs reflecting more efficient reprogramming of donor cells, even though cell numbers were not affected (Table 1). Our results are in agreement with those reported recently by Xu et al. (2012), indicating a distinct advantage of using VPA in bovine SCNT for improved embryo production and efficient reprogramming. However, we have used a different approach in this study because the somatic cells were subjected to VPA treatment prior to SCNT production, whereas in the study done by Xu et al. (2012) treatments were on the reconstructed embryos. The advantage of our approach over Xu et al. (2012) was to provide a pre-reprogrammed donor genome to the recipient oocyte so that the factors present in the oocyte cytoplast would be able to work efficiently, resulting in proper reprogramming of donor cells.
In addition to DNA methylation, covalent modifications of histones, such as acetylation, methylation, phosphorylation, and ubiquitination, also play critical roles in regulation of gene expression and are involved in the processes of epigenetic reprogramming (Li, 2002; Reik, 2007). Modifications can occur at several amino acid residues, of which the lysine residue 9 of histone H3 (H3K9) can be either acetylated or methylated. In general, acetylated H3K9 represents gene transcription active status while methylated H3K9 represents gene silencing (Jenuwein and Allis, 2001). In agreement with the other studies, our results also indicate that cloned blastocysts are generally hypermethylated on H3K9me3 in comparison to IVF-derived blastocysts (Bourc'his et al., 2001; Dean et al., 2001; Kang et al., 2001; Santos et al., 2003; Tian et al., 2009). We found that H3K9me3 methylation of cloned blastocysts derived from VPA-treated cells was decreased to the level of the IVF counterpart, even though the H3K9me3 status of donor cells was not affected by VPA (Figs. 6 and 7). We also found that parthenogenetic blastocysts had the highest level of H3K9me3 methylation among all groups. H3K9 methylation and DNA methylation are associated with silenced chromatin, and there appears to be interplay between the two epigenetic modifications (Jackson et al., 2002; Soppe et al., 2002; Tamaru et al., 2001). It is still an open question whether the effect of VPA on H3K9 methylation directs DNA methylation or the other way around. For both of these scenarios, the experimental evidence needs to be explored further.
In conclusion, this study provides strong evidence that treatment of bovine skin fibroblasts with VPA results in improved SCNT blastocyst production, with lower levels of apoptosis and H3K9 methylation, similar to those of embryos derived from IVF. We have demonstrated that VPA is an efficient drug for in vitro development of cloned embryos and enhancing nuclear reprogramming.
Footnotes
Acknowledgments
This work was funded by Canadian Institutes of Health Research (CIHR) and National Science and Engineering Research Council of Canada (NSERC). We thank Mr. Ed Reyes and Ms. Esther Semple for their technical support. N.L.S. was the recipient of a Canadian Commonwealth Fellowship Program.
Author Disclosure Statement
The authors declare that no conflicting interests exist.
References
Supplementary Material
Please find the following supplemental material available below.
For Open Access articles published under a Creative Commons License, all supplemental material carries the same license as the article it is associated with.
For non-Open Access articles published, all supplemental material carries a non-exclusive license, and permission requests for re-use of supplemental material or any part of supplemental material shall be sent directly to the copyright owner as specified in the copyright notice associated with the article.