
Abstract
Abstract
The significance of metabolic networks in guiding the fate of the stem cell differentiation is only beginning to emerge. Oxidative metabolism has been suggested to play a major role during this process. Therefore, it is critical to understand the underlying mechanisms of metabolic alterations occurring in stem cells to manipulate the ultimate outcome of these pluripotent cells. Here, using P19 murine embryonal carcinoma cells as a model system, the role of mitochondrial biogenesis and the modulation of metabolic networks during dimethyl sulfoxide (DMSO)-induced differentiation are revealed. Blue native polyacrylamide gel electrophoresis (BN-PAGE) technology aided in profiling key enzymes, such as hexokinase (HK) [EC 2.7.1.1], glucose-6-phosphate isomerase (GPI) [EC 5.3.1.9], pyruvate kinase (PK) [EC 2.7.1.40], Complex I [EC 1.6.5.3], and Complex IV [EC 1.9.3.1], that are involved in the energy budget of the differentiated cells. Mitochondrial adenosine triphosphate (ATP) production was shown to be increased in DMSO-treated cells upon exposure to the tricarboxylic acid (TCA) cycle substrates, such as succinate and malate. The increased mitochondrial activity and biogenesis were further confirmed by immunofluorescence microscopy. Collectively, the results indicate that oxidative energy metabolism and mitochondrial biogenesis were sharply upregulated in DMSO-differentiated P19 cells. This functional metabolic and proteomic study provides further evidence that modulation of mitochondrial energy metabolism is a pivotal component of the cellular differentiation process and may dictate the final destiny of stem cells.
Introduction
D
Metabolism is the foundation of any cell and usually displays the end result of gene expression and function. The importance of metabolism in dictating the final destination of stem cells is only now becoming apparent (Folmes et al., 2012; Rafalski and Brunet, 2011; Yusuf and Scadden, 2012). There is mounting evidence supporting the implication of oxidative phosphorylation in the process of differentiation (Chung et al., 2007; Yanes et al., 2010). The essential nature of energy production via oxidative metabolism for the functional excitation–contraction in human cardiac lineage was recently demonstrated (Chung et al., 2007). Natural metabolites associated with oxidative metabolism in the mitochondria have also been shown to substantially increase the level of neuronal and cardiac differentiation in human embryonic stem cells (ESCs) (Yanes et al., 2010). In addition, oxidative stress caused by oxidative phosphorylation has been reported to provide cues for differentiation (Ji et al., 2010; Schmelter et al., 2006). For instance, the activation of nicotinamide adenine dinucleotide phosphate (NADPH) oxidase and the high level of reactive oxygen species (ROS) found within the differentiating cells indicate a possible role of an oxidative environment and ROS in signaling, proliferation, and differentiation (Sauer et al., 2001). NADPH oxidase and ROS generation are two critical factors in cardiac differentiation of murine ESCs (Sauer and Wartenberg, 2005).
The role of oxidative metabolism and promotion of an oxidative environment point to the participation of mitochondria in the developmental process of stem cells. Particularly, recent studies revealing the additional functions of mitochondria make this organelle an ideal subject of investigation (Lonergan et al., 2007; Parker et al., 2009). Low numbers of mitochondrial DNA (mtDNA) have been reported in undifferentiated cells (Facucho-Oliveira et al., 2007; St. John et al., 2005). The structural dynamics of mitochondria, such as morphological maturity and mitochondrial fusion and fission, have also been found to have an impact on mammalian stem cells as well (Chen et al., 2003; Mandal et al., 2011). Despite these findings, functional studies on mitochondrial metabolism in stem cells have yet to be fully explored.
In this report, the mesoderm and endoderm differentiating agent DMSO is used to demonstrate the altered mitochondrial metabolism in P19 embryonal carcinoma cells. This functional metabolic study provides evidence for the elevated energy-producing ability of the differentiated P19 cells. Mitochondrial biogenesis appears to be a key modulator of these transforming cells. The significance of metabolism in dictating the destiny of pluripotent cells is also discussed.
Material and Methods
Cell culture
The P19 ESCs were obtained from the American Type Culture Collection (ATCC #CRL-1825). The cells were propagated in the absence of feeder layers in Falcon 75-cm2 vented cap tissue culture flasks. The cells were cultured in α-minimum essential media (αMEM; Fisher Scientific) supplemented with 10% fetal bovine serum (FBS) and 1% antibiotics. The cells were rinsed with phosphate-buffered saline (PBS; 136 mM sodium chloride, 2.5 mM potassium chloride, 1.83 mM dibasic sodium phosphate, and 0.43 mM monobasic potassium phosphate, pH 7.4) and incubated with trypsin (1×) in PBS for detachment. A 1:9 dilution was used to seed the new population. All cultures were kept at 5% CO2 and 37°C humidified atmosphere (Gong et al., 2013).
Differentiation and cell fractionation
To induce differentiation, 1×106 cells were plated in 10 mL of αMEM containing 1% DMSO. The control population was cultured in the absence of DMSO. The cells were transferred into 10-cm petri dishes consisting of a thin layer of agar to prevent adhesion. After 2 days, the original medium was carefully removed and replenished with fresh medium. On the fourth day, embryoid bodies were trypsinized for disaggregation and centrifuged at 800×g for 10 min at 4°C. An equal number of cells from both control and DMSO-differentiated cultures was collected and suspended in the cell storage buffer (CSB; 50 mM Tris-HCl, 1 mM phenylmethylsulfonylfluoride, 1 mM dithiothreitol, 250 mM sucrose, and 2 mM citrate). The cells were fractionated into nuclear, mitochondrial, and cytoplasmic portions as described in Lemire et al. (2008b). The Bradford assay was used to quantify the protein content from nuclear, mitochondrial, and cytoplasmic fractions. Bovine serum albumin (BSA; Sigma Aldrich) was used as the standard (Bradford, 1976).
Blue native-polyacrylamide gel electrophoresis and in-gel activity assay
Blue native polyacrylamide gel electrophoresis (BN-PAGE) was performed as described in Han et al. (2012) and Schägger and von Jagow (1991). The 4–16% gradient gel was used for the protein separation, and the BioRad MiniProtein™ 2 system was used for electrophoresis. Mitochondrial and nuclear proteins were prepared in BN buffer (500 mM 6-aminohexanoic acid, 50 mM BisTris, pH 7.0) and 1% β-dodecyl-
Western blot analysis
Sodium dodecyl sulfate (SDS)-PAGE was performed with the modified method as described (Laemmli, 1970; Singh et al., 2005). The proteins were subjected to 62.5 mM Tris-HCl (pH 6.8), 2% SDS, and 2% β-mercaptoethanol at 100°C for 5 min. The proteins were separated with 10% isocratic denaturing gel. Two-dimensional (2D) immunoblotting was performed as previously described (Lemire et al., 2011). The proteins were blotted onto a nitrocellulose membrane overnight at 4°C. After the membrane was treated with 5% nonfat skim milk in Tween-20 Tris-buffered saline (TTBS; 20 mM Tris-HCl, 0.8% NaCl, 1% Tween-20, pH 7.6) to prevent nonspecific binding for an hour, it was incubated overnight with the primary antibody OCT4 (1:800, Abcam). The membrane was subsequently washed with TTBS and incubated with secondary antibody for an hour before visualization. Fluorescent-tagged secondary antibodies (1:10,000) (Li-Cor, Lincoln, NE, USA) allowed for detection using the Odyssey infrared imaging system (Li-Cor).
Immunofluorescence microscopy
The cells were collected and disaggregated on day 4 of the differentiation protocol. The cells were allowed to adhere to the coverslip overnight at 5% CO2 and 37°C humidified atmosphere. Following the wash (PBS containing 0.5 mM EDTA), the slides were first incubated in Hoechst 33528 (2.5mu;g/mL in PBS) and then subjected to Rhodamine B (15 μg/mL in α-MEM) for 20 min. This dye is known to respond to mitochondrial membrane potential (Lemire et al., 2009). The cells were washed with PBS and submerged in TTBS with 5% FBS for an hour to block nonspecific binding sites. Tris-buffered saline (TBS) was used for rinsing, and the slides were introduced to voltage-gated anion channel (VDAC) antibody (1:500 Abcam) and peroxisome proliferator-activated receptor-γ coactivator 1-α (PGC-1α) antibody (1:500 Abcam) at room temperature. The coverslips were then washed thoroughly with TBS and incubated with secondary antibody conjugated to fluroescein isothiocyanate (FITC; 1:1000, Sigma Aldrich). A Zeiss inverted deconvolution microscope was used to observe the cellular morphology of the cell.
Metabolite analysis
Metabolite and ATP levels were monitored by HPLC. The whole-cell lysates were used to analyze the metabolite profiles. Equal numbers of cells were used in all experiments. Intact mitochondrial fractions were incubated in 5 mM succinate or 5 mM malate in presence of adenosine diphosphate (ADP) for an hour. The samples were injected into a C18 reversed-phase column (Synergi Hydro-RP, Phenomenex) operating at a flow rate of 0.7 mL/min at ambient temperature. KH2PO4 (20 mM, pH 2.9) was used as a mobile phase to separate organic acids, which were then detected using a Waters dual-absorbance detector at 210 nm. Nucleic acids were detected at 254 nm. Metabolites were identified using known standards, and the samples were spiked with the standards to confirm the peak retention times. Quantification of the peaks was performed by the Empower software (Waters Corporation).
Statistical analysis
All experiments were performed three times and in duplicate. Where appropriate, the Student t-test was used to assess significance.
Results
Metabolite profile of differentiating P19 cells
Prior to performing any experiment, OCT4 expression was monitored to ensure whether the DMSO indeed induced differentiation of P19 cells. A markedly low expression of OCT4 in DMSO-treated cells suggests that DMSO was indeed an efficient morphogen for this cell line (Fig. 1a). To examine the modification in metabolism during DMSO-induced differentiation, the colony-forming efficiency (CFE) of the undifferentiated and differentiated cells was subjected to HPLC analysis. The comparison between undifferentiated (control) and differentiated (DMSO-treated) cells at day 4 revealed significant changes in metabolite levels (Fig. 1b). Differentiated cells isolated at day 3 and day 7 also indicated similar trends, displaying drastically altered metabolite profiles (Fig. 1c). TCA intermediate metabolites, such as pyruvate, isocitrate, α-ketoglutarate, and succinate, were sharply increased. Lactate content, however, appeared significantly reduced in the differentiated cells.

Metabolite profile of differentiating stem cells. (
Metabolic regulation and differentiation
BN-PAGE activity assays demonstrated changes in activities of various key central metabolic enzymes during differentiation. Glycolytic enzymes, HK, GPI, and pyruvate kinase (PK) displayed increased activity proceeding from day 3 to day 7, thus suggesting enhanced glycolysis during DMSO-induced differentiation (Fig. 2a). Oxidative phosphorylation was analyzed by monitoring the activity of Complex I and Complex IV. These two electron transport chain (ETC) complexes indeed exhibited a higher level of activity, an indication of enhanced oxidative energy metabolism in the cell (Fig. 2b). Enzymatic activity was compared to the undifferentiated counterpart to ensure that the alterations were indeed a result of differentiation promoted by the presence of DMSO (Fig. 2c). The changes in metabolic profiles and enzymatic activities pointed to increased mitochondrial activity. To confirm the upregulation of mitochondrial activity, mitochondrial ATP production and membrane potential of the mitochondria were assessed. The level of mitochondrial ATP production was monitored via HPLC to further solidify this hypothesis. TCA intermediates, succinate and malate, and ADP were added to the mitochondrial fractions. In both cases, differentiated cells were able to produce ATP at a higher rate compared to the undifferentiated cells (Fig. 3a). Fluorescent microscopy using rhodamine dye also confirmed the presence of enhanced membrane potential in the DMSO-treated cells, thus pointing to elevated mitochondrial activity (Fig. 3b).

Metabolic enzyme activity in DMSO differentiated cells. (
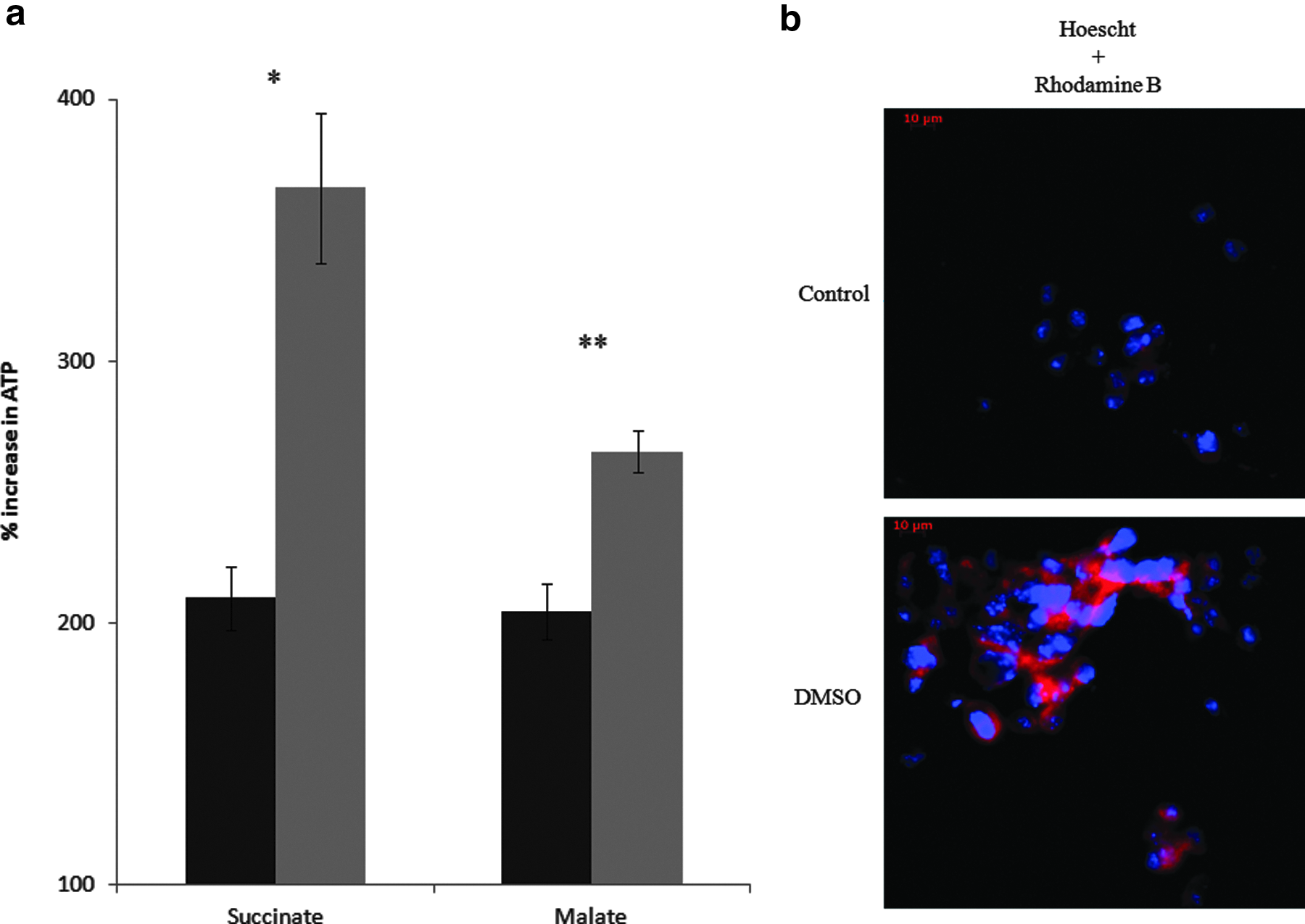
Mitochondrial activity in DMSO-differentiated cells. (
Mitochondrial biogenesis and metabolism in the early stage of differentiating cells
Mitochondrial biogenesis was examined by assessing the level of VDAC and PGC-1α expression. VDAC is a mitochondrial marker; thus, VDAC expression should be elevated if the differentiated cells consist of higher mitochondrial content. The results indicated that DMSO-treated cells at day 4 had increased level of VDAC (Fig. 4a). The merged image of VDAC and rhodamine displayed intense fluorescence, an observation attributed to higher mitochondrial activity coupled to increased mitochondrial biogenesis. The level of mitochondrial biogenesis was further confirmed by examining the expression of PGC-1α, a master regulator of mitochondrial biogenesis. Differentiated cells consisted of markedly increased fluorescence, characteristic of this important metabolic modulator (Fig. 4b).
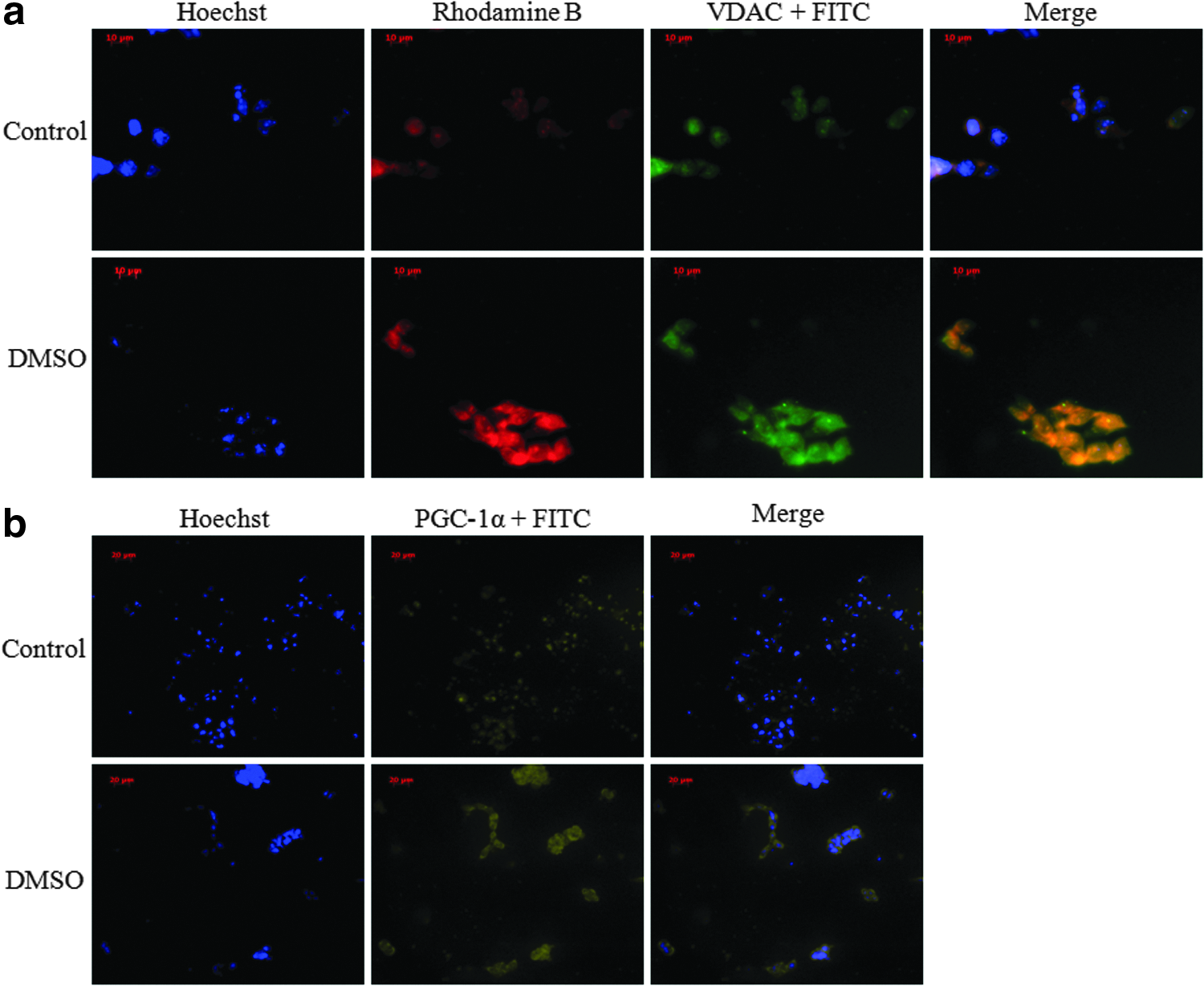
(
Discussion
Upon treatment with DMSO, the CFE of P19 cells demonstrated a metabolic profile that was in sharp contrast to the control. The peaks attributed to pyruvate, isocitrate, α-ketoglutarate, and succinate were found to be significantly elevated in the CFE of the DMSO-treated cells (Fig. 1b,c). The higher level of lactate in the untreated cells indicates that these cells employed anaerobic substrate-level phosphorylation to generate energy, an observation common in undifferentiated cells (Fig. 1b,c) (Chung et al., 2007; Varum et al., 2011). The DMSO-treated cells, however, consisted of an increased level of TCA cycle metabolites like α-ketoglutarate and succinate, indicative of pronounced oxidative metabolism (Fig. 1b,c). Hence, it was evident that the metabolic networks in the cells were undergoing major alterations to accommodate the differentiation process. The decrease in OCT4, a marker for differentiating cells (Niwa et al., 2000) in the DMSO-treated P19 cells did confirm that these cells were losing their pluripotency (Fig. 1a).
To further elucidate the disparate products observed in these cultures, various enzymes promoting the formation of pyruvate, a key substrate for oxidative energy production, were assessed. Cytosolic enzymes such as HK, GPI, and PK had increased activities as the cells continued maturation postdifferentiation (Fig. 2a). Because the lactate production level was found to be lower and the pyruvate content higher in the differentiated cells, it was clear that glycolysis was providing pyruvate to fuel the TCA cycle (Fig. 1b,c). Pyruvate can then enter the TCA cycle and ultimately promote ATP production via oxidative phosphorylation. This is the first demonstration of the increased activities of glycolytic enzymes supplying pyruvate for enhanced production of energy in the DMSO-treated differentiating cells. To prove if indeed the destination of pyruvate was the ATP-generating machinery of the cell, Complex I and Complex IV were monitored. As anticipated, the activities of these enzymes were markedly upregulated (Fig. 2b). Although the activation of mitochondrial function via the formation of ROS and upregulation of antioxidants like catalase and glutathione has been shown (Cho et al., 2006), this is the first demonstration of the direct participation of oxidative phosphorylation during DMSO-induced differentiation. The upregulation of both key ETC enzymes provides strong evidence that mitochondrial metabolism was sharply modified under DMSO-induced differentiation conditions and that the energy needs of these cells are different from the pluripotent cells. All enzymatic activity assays were performed on control proteins to visualize the contribution of spontaneous differentiation (Fig. 2c).
Because the activity of the mitochondrial enzymes and the energy-generating machinery were affected by the morphogen DMSO, it was important to discern if these observations were coupled with increased ATP production. The higher concentration of metabolites such as isocitrate, α-ketoglutarate and succinate is indicative of active mitochondrial metabolism (Fig. 1a,b), thus suggesting increased ATP production. The mitochondrial fractions were incubated with TCA cycle substrates, succinate and malate, in the presence of ADP. Indeed, higher ATP production in the mitochondria was observed (Fig. 3a). Hence, it was evident that the differentiating cells had higher oxidative phosphorylation capacity than the stem cells. The mitochondrial activity was also detected by examining the membrane potential via Rhodamine B dye (Fig. 3b). Mitochondria are known to convert transmembrane potential into ATP, thus it would be higher in the treated cells. Indeed, Rhodamine B- and VDAC-attributed fluorescence were markedly intense in the differentiating cells (Fig. 4a). The localization of mitochondria in differentiated cells was also shown to undergo extensive distribution throughout the cells, as described previously (Cho et al., 2006; Mandal et al., 2011). Because there was more need for ATP generated by oxidative phosphorylation, it is logical that mitochondrial biogenesis would be promoted in the differentiating cells. The increased immunofluorescence observed with VDAC was coupled to the upregulation of PGC-1α, a transcription factor known to promote the synthesis of mitochondria (Fig. 4b) (Scarpulla, 2011).
In summary, this study provides a functional state of the metabolic module of the DMSO-induced differentiating P19 embryonal carcinoma cells. Upon treatment with DMSO, the metabolic networks undergo a sharp reprogramming. The increase in mitochondrial biogenesis is coupled to the upregulation of the activities of various enzymes involved in ATP formation via oxidative phosphorylation. This functional metabolic study reveals the critical role metabolism plays in guiding cellular differentiation and lays the groundwork for the development of technologies aimed at the manufacture of desired cell types.
Footnotes
Acknowledgment
This work has been funded by Laurentian University. Christopher Auger is a recipient of the NSERC postgraduate scholarship. Sean C. Thomas is a recipient of the Ontario Graduate Scholarship (OGS).
Author Disclosure Statement
The authors state that there are no conflicts of interest and they have received no payment for preparation of this manuscript.