
Abstract
Abstract
Induced pluripotent stem cells (iPSCs) are usually generated by reprogramming somatic cells through transduction with a transcription factor cocktail. However, the low efficiency of this procedure has kept iPSCs away from the study of the clinical application of stem cell biology. Our research shows that continuous passage increases the efficiency of reprogramming. Compared with conventional method of establishment of iPSCs, more embryonic stem cell (ESC)-like clones are generated by continuous passage during early reprogramming. These inchoate clones, indistinguishable from genuine ESC clones, are closer to fully reprogrammed cells compared with those derived from classical iPSC induction, which increased the expression of pluripotent gene markers and the levels of demethylation of Oct4 and Nanog. These results suggested that full reprogramming is a gradual process that does not merely end at the point of the activation of endogenous pluripotency-associated genes. Continuous passage could increase the pluripotency of induced cells and accelerate the process of reprogramming by epigenetic modification. In brief, we have provided an advanced strategy to accelerate the reprogramming and generate more nearly fully reprogrammed iPSCs efficiently and rapidly.
Introduction
S
Many studies have shown that the inhibition of the p53/p16 pathway enhances the generation of iPSCs, and a high cell division rate accelerates the process of direct reprogramming slightly (Banito et al., 2009; Gao et al., 2013; Lin and Ying, 2012; Smith et al., 2010). Indeed, our research found that continuous passage of mouse iPSCs not only upregulated pluripotent genes, but also resulted in a highly increased demethylated change in the promoters of both Oct4 and Nanog. Thus, continuous passage during the early stage could significantly increase the pluripotency of iPSCs with epigenetic modification.
Materials and Methods
Cell culture
Mouse embryonic fibroblasts (MEFs) were isolated from E13.5 B6D2F1 mouse embryos and washed in phosphate-buffered saline (PBS). The head and visceral tissues were removed from isolated embryos. The remaining bodies were washed in fresh PBS, minced to 1- to 3-mm pieces using a pair of scissors, transferred into a tube with 0.1 mM trypsin/1 mM EDTA solution, and incubated for 3 min. After incubation, Dulbecco's modified Eagle medium (DMEM) with 10% fetal bovine serum (FBS) was added to stop trypsinization, and the supernatant was transferred into a new tube. Cells were resuspended in fresh medium and cultured on 100-mm dishes at 37°C with 5% CO2.
R1 cells were cultured in DMEM medium with 15% embryonic cell–qualified FBS (ES-FBS; Invitrogen), 0.1 mM nonessential amino acids (NEAA; Invitrogen), 0.1 mM β-mercaptoethanol, and 1000 U/mL leukemia inhibitory factor (LIF; Gibco) and passaged every other day. Plat-E packaging cells (Cyagen), which were used to produce retroviruses, were maintained in DMEM medium containing 10% FBS, 50 mg/mL penicillin/streptomycin, 1 mg/mL puromycin (Sigma), and 100 mg/mL of blasticidin S (Merck).
Establishment and in vitro differentiation of iPSCs
Retroviral production was performed as described by Yamanaka with minor modifications. Plat-E cells were seeded at 8×106 cells per 100-mm dish. The following day, pMXs-based retroviral vectors of Oct4, Sox2, Klf4, and c-Myc were introduced into Plat-E cultures using Lipofectamine® LTX & PLUS transfection reagent (Invitrogen). Virus-containing supernatants were collected 48 h later and filtered through a 0.45- μm cellulose acetate filter (Millipore) and supplemented with 4 mg/mL Polybrene (Sigma). Equal amounts of supernatants containing each of the four retroviruses were transferred to the fibroblasts dish and incubated overnight. On the next day, MEFs were infected again. After 24 h (labeled day 0), the medium was replaced with MEF medium. After the infected MEFs were seeded on mitomycin C–treated MEFs, the medium was changed to knockout DMEM medium with 15% Knockout Serum Replacement (KOSR; Invitrogen), 0.1 mM NEAA, 0.1 mM β-mercaptoethanol, and 1000 U/mL LIF. The ESC-like clones were picked and amplified.
After the generation of iPSCs, the cells were harvested by trypsinization and transferred to low-attachment Petri dishes in the ESC medium without LIF. After 4 days, aggregated cells were plated onto gelatin-coated tissue culture dishes and incubated for another 4 days. The cells were stained with germ layer antibody [Tuj1, smooth muscle actin [SMA], α-fetoprotein (AFP)].
Teratoma formation assay
iPSCs were suspended at 1×107cells/mL in PBS. A 100- μL amount of the cell suspension was used for subcutaneous injection into the dorsal flank of nonobese diabetic–severe combined immunodeficient (NOD-SCID) mice. Four weeks after the injection, tumors were surgically dissected from the mice, fixed with 4% paraformaldehyde, and embedded in paraffin. Sections were stained with Hematoxylin & Eosin.
Alkaline phosphatase and immunofluorescent staining
For alkaline phosphatase (AKP) staining, cells were first washed with PBS, fixed in 4% paraformaldehyde (PFA), and washed again with PBS. The cells were stained with AKP solution according to the manufacturer's guidelines (Beyotime Institute of Biotechnology).
For immunocytochemistry, cells were fixed with 4% PFA for 30 min, permeabilized with 1% Triton X-100 for 15 min, and blocked with 2% goat serum in PBS for 1 h. Primary antibodies used were anti-Oct3/4 (Abcam), anti-stage-specific embryonic antigen-1 (SSEA1) (Santa Cruz), anti-AFP (Boster), anti-Tuj1 (Santa Cruz), and anti-SMA (Boster). After incubation overnight with primary antibodies, cells were washed with PBS twice and incubated with secondary antibodies (Santa Cruz). After washing with PBS, the samples were counterstained with Hoechst 33258 (Invitrogen). Fluorescent images were taken under a Nikon microscope using appropriate filters.
RT-PCR and real-time PCR analysis
Total RNA was isolated with TRIzol and treated with DNase (Promega). RNA was reverse transcribed using a High Capacity cDNA Reverse Transcription Kit (ABI, cat. no. 4368814). PCR cycling parameters were 5 min at 94°C followed by thirty cycles of 30 sec at 94°C, 30 sec at 60°C, 30 sec at 72°C, and a final 10-min extension at 72°C. PCR products were separated on a 2% agarose gel, stained with ethidium bromide, visualized, and photographed on a UV transilluminator. Real-time PCR reactions were performed based on a 1- μL cDNA sample, 10 μL of TransStart™ Top Green qPCR SuperMix (TransGen, AQ131), and gene-specific primers in a 20- μL reaction system on CFX96 Real-Time System (Bio-Rad, USA). Each sample was analyzed in triplicate with glyceraldehyde 3-phophate dehydrogenase (Gapdh), H2a, and β-actin as the internal controls. Primer sequences used in this study are shown in Table S1 (Supplementary Data are available at www.liebertpub.com/cell/.) The Student's t-test was used to assess differences between groups. p<0.05 was considered statistically significant.
Bisulfite genomic sequencing
Bisulfite treatment was performed using an EZ DNA Methylation Kit (Zymo Research) according to the manufacturer's recommendations. Amplified products were cloned into pCR2.1-TOPO (Invitrogen). Ten randomly selected clones were sequenced with the M13 forward and M13 reverse primers for each gene. We obtained PCR primer sequences for SSLP from the Mouse Genome Informatics website. Allele sizes were approximated on the basis of the known allele sizes in various inbred strains.
Results
Passaging culture increased the generation of ESC-like clones
We infected B6D2F1 MEFs by retrovirus with cDNAs carrying four Yamanaka factors as described in the Material and Methods section. Infected MEFs were transferred onto feeder layers at day 3 after infection and cultured in ESC medium (Fig. 1Aa). Cell groups with an epithelium-like morphology first emerged at day 6 (Fig. 1Ab). ESC-like colonies were large and round with clear boundaries at day 8 (Fig. 1Ac). At day 10, cells with different morphology filled the plate and included ESC-like populations, a separate population of small, round, rapidly dividing cells (Fig. 1Ad). Instead of picking the clones, we passaged the cells as an entire plate, which gave rise to a heterogeneous culture.
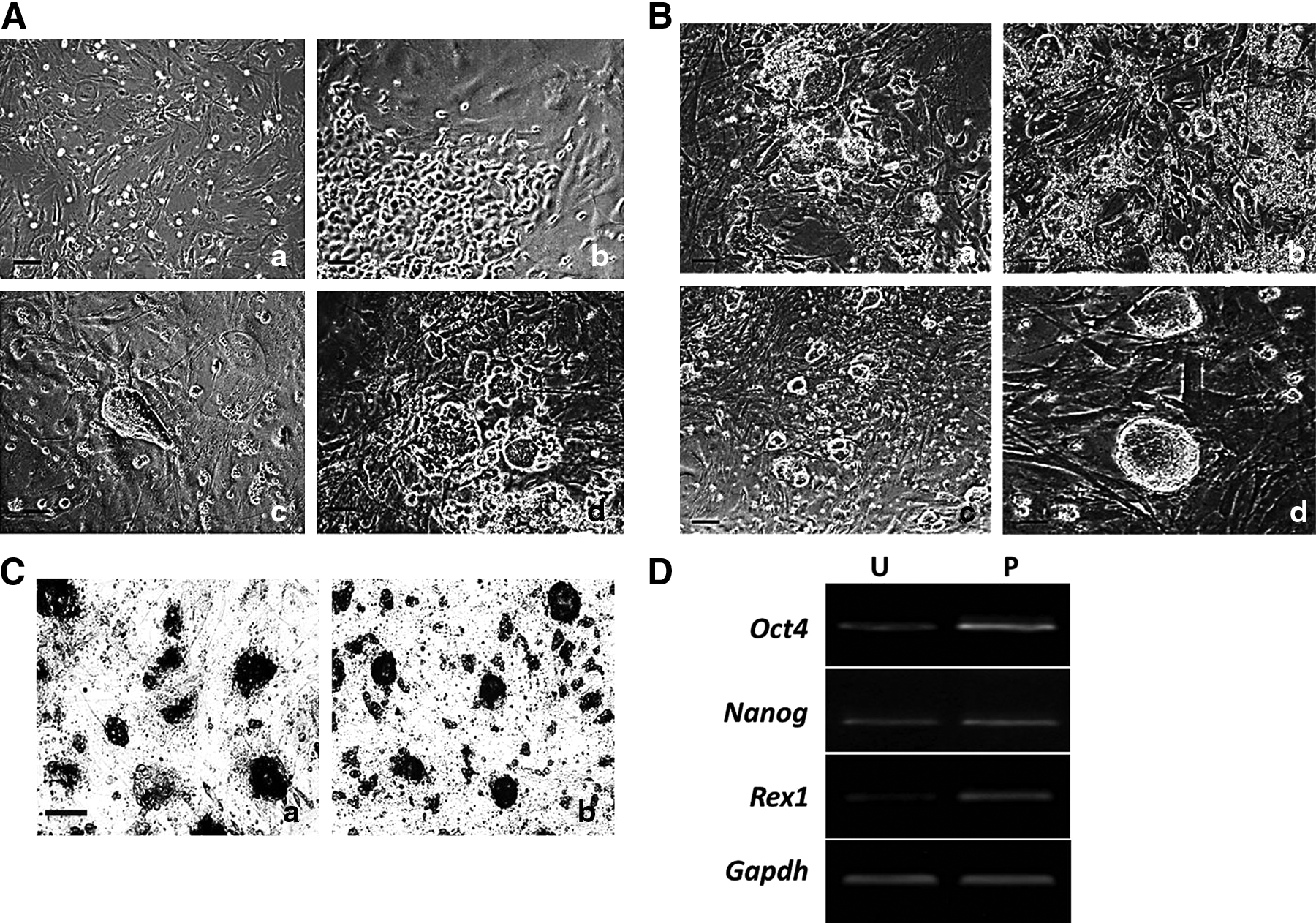
Generation of miPSCs cells by heterogeneous culture. (
After several passages, it turned out that the more cells were passaged, the more numbers of ESC-like colonies were produced in heterogeneous culture cells (Fig. 1B). The heterogeneous culture clones exhibited more multicellular, clearly marginal, and regular morphology (Fig. 1Bd). AKP staining showed that more AKP-positive cells were found in heterogeneous cultured cells compared with unpassaged cells (Fig. 1C). RT-PCR results showed the expression of pluripotent gene markers, such as Oct4, Nanog, and Rex1, were higher in heterogeneous cultured cells than that in unpassaged induced cells (Fig. 1D).
Afterward, we randomly picked 10 ESC-like colonies from unpassaged cells and heterogeneous cultured cells by morphological criteria, and then generated 6 or 10 iPSCs, respectively. Some clones derived from unpassaged cells did not form iPSCs by morphological criteria. The efficiency of induction by heterogeneous culture was significantly higher than in unpassaged cultures. These results suggested cell passage might play an important role in early MEF reprogramming.
Identification of unpassaged and passaged cultured iPSCs
B4 iPSCs cells derived from unpassaged cultured cells and C1 iPSCs from passaged (also called heterogeneous) cultured cells were randomly chosen for further identification. Both B4 and C1 iPSCs had been stably expanded for over 30 passages and maintained a normal karyotype. The two iPSCs lines were morphologically indistinguishable from classic mouse ESCs, displayed typical ESC-like morphology, including a high nucleus-to-cytoplasm ratio, and had clear boundaries (Fig. 2A). They both expressed the typical ESC marker genes Oct4 and Ssea1 (Fig. 2B). After culture in differentiated medium for 8 days, the two iPSC lines expressed the three germ layer markers, Tuj1 (ectoderm), SMA (mesoderm), and AFP (endoderm), as seen by by immunofluorescence (Fig. 2C). Furthermore, the two iPSC lines were injected subcutaneously into nude mice; after 4 weeks, this resulted in teratomas comprising tissues derived from the three germ layers (Fig. 2D). These results showed that the pluripotency of iPSCs was indistinguishable after longer passage.

miPSCs are generated from unpassaged and passaged cultured cells. (
Continuous passage affects the pluripotency of induced cells
All analysis above suggested that passage could accelerate the process of reprogramming when early iPSCs cells were being established. To verify the effect of passage on the reprogramming, we divided the induced cells into two groups after infection. One group (UN, n=5) was passaged every 3 days after picking ESC-like clones, and the medium was changed every day; the other group (PA, n=5) was passaged after picking ESC-like clones every day. Although all colonies were cultured in the same medium, the morphology of clones in the PA group was different from the UN group after 3 days. All clones of PA group exhibited a round surface with a clear margin whereas some clones of the UN group remained flat or separated into a population of small round cells (Fig. 3A). After 6 days of culture, we examined the expression levels of several pluripotent genes in two groups by real-time PCR. Endogenous Oct4, Sox2, Nanog, Rex1, and E-cadherin were upregulated in the PA group compared with the UN group (Fig. 3B). Afterward, we investigated DNA methylation status at the promoter regions of Oct4 and Nanog in two groups by bisulfite genomic sequence analysis. The data demonstrated that the levels of methylation in the promoter regions of Oct4 and Nanog were decreased during the reprogramming. The extent of demethylation of Oct4 in the PA group was more significant than that of Nanog, which was becoming similar to that in R1 cells (Fig. 3C). However, after culturing for passage 30, both Oct4 and Nanog were nearly absolutely demethylated (Fig. 3C). In this case, continuous passage of miPSCs could promote the expression of pluripotent genes by epigenetic modification.
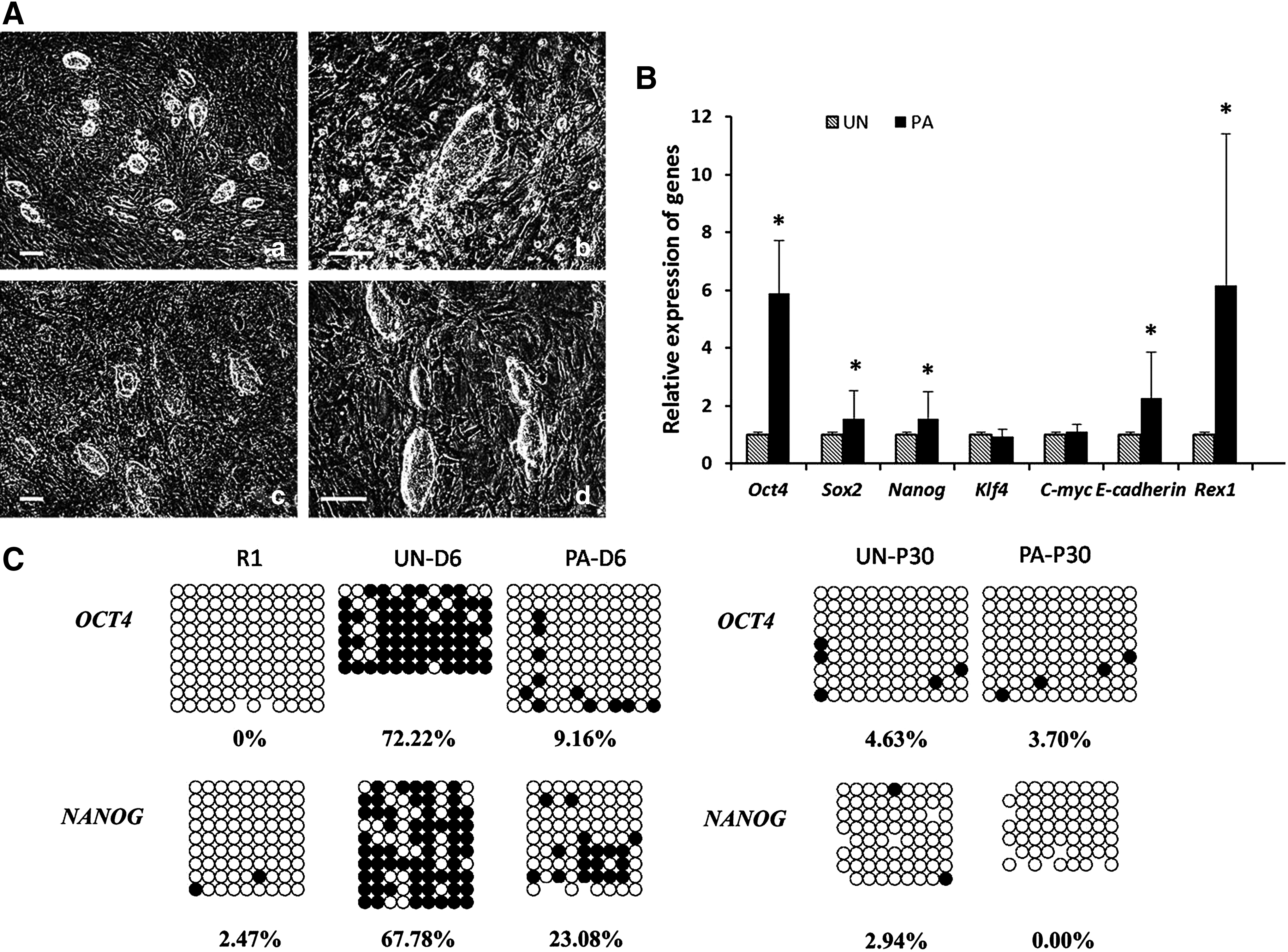
Comparison of unpassaged and continuously passaged cells during early reprogramming. (
Discussion
Previous studies on iPSCs had demonstrated their application not only in the study of stem cell biology, but also in clinical utility (Gandre-Babbe et al., 2013; Hunt, 2011; Wichterle and Przedborski, 2010). However, low efficiency was a significant barrier for generation of iPSCs. Notably, some other reports identified that cellular origin influences the in vitro differentiation potentials of early-passage iPSCs into the other cell types. Correspondingly, it takes a long time for the complete reprogramming converting MEFs to iPSCs. Our study showed rapid upregulation of pluripotent genes by continuous passage of miPSCs during early reprogramming, which suggested that full reprogramming was a gradual process that did not merely end at the point of the activation of endogenous pluripotency-associated genes. The upregulated genes included Oct4, Sox2, and Nanog during continuous-passage culture, especially the high expression of Oct4. There was no significant difference in the expression of c-Myc between the PA group and the UN group. This is in accordance with the previous report that c-Myc is at a low level at the end of reprogramming process, and that it plays a vital role as transcriptional activator (Ohi et al., 2011).
During the process of induction, we found that it was difficult to generate 100% iPSCs by only morphological selection because some ESC-like clones may not be genuine fully reprogrammed cells, which would be time-consuming and a waste of money. However, if the ESC-like clones were picked after culturing for several passages, almost all would generate fully reprogrammed iPSCs; this increased the efficiency of establishing these cells. This suggested that the early passage could accelerate the generation of pluripotency. To verify this hypothesis, we detected the effect of passage on the pluripotency. The results showed that compared with the long-time passage (passage each 3 days), the morphology of induced cells was closer to that of ESCs with clear borders. Molecular analysis showed continuous passage increased the expression of pluripotent gene markers, such as Oct4 and Nanog, which were key genes involved in the regulated network of ESCs. However, pluripotent expression of these genes became similar after long passage (>10 passages). This was in accordance with what was reported previously, that the transcriptional pattern in early-passage iPSCs cells is different from the transcriptional pattern in late-passage iPSCs (Barrero and Izpisua Belmonte, 2011; Lister et al., 2011). This suggested that the effect of passage on pluripotency occurred during the early reprogramming. Furthermore, we investigated the regulatory mechanism of continuous passage. We found that continuous passage increased the level of demethylation of Oct and Nanog, demonstrating that the passage regulates the expression of pluripotent gene markers by epigenetic modification.
Although the induction of iPSCs is a long and unstable process, it can be modified by other methods, such as changing the vectors carrying transcription factors or adding small molecules in induced cells (Mochiduki and Okita, 2012). The research reported here revealed that continuous passage could accelerate reprogramming by increasing the pluripotency in early induced cells, thus providing an advanced strategy for rapid iPSCs generation. Interestingly, we wondered whether partially reprogrammed iPSCs could turn into fully-reprogrammed iPSCs by continuous passage. Future research will focus on this aspect.
In conclusion, our study showed that continuous passage could affect the pluripotency of induced cells and accelerate the process of reprogramming by promoting transcriptional reactivation and modifying epigenetic inheritance. This is a meaningful tool for studying the mechanism of reprogramming of somatic cells.
Footnotes
Acknowledgments
The authors are grateful to the laboratory of Professor Liu at North East Agricultural University for their valuable technical assistance. This article is funded by the National Natural Science Foundation of China (grant no. 31000645), the National Natural Science Foundation of China (grant no. 30900413), Ph.D. Programs Foundation of Ministry of Education of China (grant no. 20092307120016 ), and The National Natural Science Heilongjiang Outstanding Youth Fund (grant no. JC200905).
Author Disclosure Statement
No competing financial interests exist.
References
Supplementary Material
Please find the following supplemental material available below.
For Open Access articles published under a Creative Commons License, all supplemental material carries the same license as the article it is associated with.
For non-Open Access articles published, all supplemental material carries a non-exclusive license, and permission requests for re-use of supplemental material or any part of supplemental material shall be sent directly to the copyright owner as specified in the copyright notice associated with the article.