
Abstract
Abstract
Induced pluripotent stem cells (iPSCs) have potential applications in the restoration of fertility, regenerative medicine, and animal biotechnology. In this study, we present the induction of iPSCs from mouse Sertoli cells (SCs) by introducing four factors—Oct4, Sox2, Klf4, and c-Myc. As early as day 3 after induction, expression of these factors was detected and typical embryonic stem-like cells began to form. On day 18, these exogenous genes were silenced and colonies were selected according to morphological characteristics. The iPSCs induced from SCs, termed SCiPSCs, strongly expressed pluripotent markers, showed a normal karyotype, and had proliferation and differentiation characteristics similar to those of embryonic stem cells (ESCs), both in vitro and in vivo. Furthermore, exposure of SCiPSCs to nitric oxide (NO) allowed them to maintain pluripotency through the activation of the pluripotent genes Oct4 and Sox2 and upregulation of Nanog expression. Moreover, NO prevented SCiPSCs from undergoing apoptosis by activating the antiapoptotic genes Bcl2 and Bcl2lll, downregulating the proapoptotic genes Bak1 and Casp7, and blocking the activation of the proapoptotic gene Bac. These effects were reversed by exposure to
Introduction
I
Sertoli cells (SCs) surround, support, nourish, and form the blood–testis barrier around spermatogonial stem cells. SCs are easier and cheaper to obtain than fetal fibroblasts, because the testes are removed from many animals as part of the castration process. In addition, the transcription factor Klf4, which can be used in the induction of iPSCs, is expressed in SCs (Behr and Kaestner, 2002), and SCs are more resistant to ultraviolet (UV)-induced DNA damage than are cells obtained from skin. Therefore, we hypothesized that SCs may be an excellent cell type from which to generate iPSCs.
Nitric oxide (NO), an intracellular signaling molecule, is synthesized by NO synthase (NOS) in vivo (Moncada and Higgs, 1993) and may affect the features of stem cells by regulating transcription factors and signaling pathways. NO is involved in cell survival and self-renewal of ESCs (Kanno et al., 2004; Mora-Castilla et al., 2010; Mujoo et al., 2008; Tejedo et al., 2010). The regulation of NO is concentration-dependent: High NO levels promote the differentiation of ESCs, whereas low levels contribute to the maintenance of pluripotency. We speculated that NO may also have influences on iPSCs.
In this study, we show that pluripotency can be induced in SCs by introduction of the four transcription factors. Furthermore, we also found that low NO levels improved the self-renewal and pluripotency of iPSCs induced from SCs (SCiPSCs), suggesting that NO can be used to maintain iPSCs. Our findings open a new way for scientists and doctors working in andrology to obtain stem cells, i.e., iPSCs.
Materials and Methods
Ethics statement
All experimental procedures were approved by the Committee for the Ethics on Animal Care and Experiments of the Chinese Academy of Agricultural Sciences. KM mice (Kunming White outbred strain) were purchased from the central animal laboratory of Peking University and housed under standard lighting (12 h light, 12 h darkness) at 24±2°C with access to food and water ad libitum.
Primary culture of SCs and embryonic fibroblasts
The modified procedure for isolation of SCs was based on previous studies (Guan et al., 2009). Briefly, the testes were removed from 5- to 6-day-old male KM mice and decapsulated. Seminiferous tubule fragments were digested with 0.1% collagenase IV at 37°C in a CO2 incubator for 20 min. The fragments were washed with phosphate-buffered saline (PBS) and, incubated with 0.25% trypsin/1 mM EDTA at 37°C for 5 min. The reaction was terminated by addition of fetal bovine serum (FBS). Cells were filtered through a 70-μm mesh and separated by discontinuous Percoll density gradient centrifugation (Morena et al., 1996). The fractions containing SCs were collected, and cells were washed three times and cultured in SC medium [10% FBS in Dulbecco's modified Eagle medium (DMEM) supplemented with 1% insulin-transferrin-selenium (ITS) and antibiotics]. SCs were used within two passages, because the proliferative capacity of SCs gradually decreases over several weeks (Vergouwen et al., 1991).
Mouse embryonic fibroblasts (MEFs) were isolated from mouse embryos between 12.5 to 13.5 days postcoitus. The head, limbs, and visceral tissues were removed, and the remains were minced and transferred into 0.25% trypsin/1 mM EDTA for digestion at 37°C for 5 min. After termination of the trypsin activity with FBS, the cells were cultured in SC medium at 37°C with 5% CO2. MEFs were used within five passages.
Identification of SCs
SCs were detected by immunocytofluorescence (ICF) using a rabbit anti-mouse vimentin antibody (Abcam, Hong Kong), a specific marker for SCs, in conjunction with fluorescein isothiocyanate (FITC)-conjugated goat anti-rabbit immunoglobulin G (IgG) as the secondary antibody. ICF performed with mouse fibroblasts was used as a control. The SC cells were further analyzed by reverse transcription-polymerase chain reaction (RT-PCR). Mouse glyceraldehyde 3-phosphate dehydrogenase (GAPDH) was used as a housekeeping gene. RT-PCR primers are listed in Table 1.
F, forward, R, reverse.
Transfection
Plat-E packaging cells were transfected with pMX-based vectors (including pMX-GFP vector) with Lipofectamine 2000 (Invitrogen, Beijing China) following the manufacturer's instructions. The retroviral particle–containing supernatants were collected at 48 h and 72 h, and filtered through 0.45-μm filters. Equal amounts of supernatants from each time point were mixed and supplemented with 6 μg/mL Polybrene. SCs at passage 2 were harvested and suspended in six-well culture dishes with the viral particle–containing medium. The transfection process was then repeated to increase efficiency. Medium containing 50 μg/mL vitamin C and KnockOut Serum Replacement (KSR) was changed daily to increase transduction efficiency (Teich et al., 1977; Wernig et al., 2007). Colonies were collected after 3–4 weeks. SCs without transfection were used as controls.
Cell culture
R1-ESCs were maintained on feeder cells in mouse ESC medium, consisting of knockout DMEM supplemented with 15% ES-qualified FBS, 1000 U/mL leukemia inhibitory factor (LIF), 2 mM
SCiPSCs were cultured with feeder cells in mouse iPSC medium, consisting of knockout DMEM supplemented with 20% KSR, 1000 U/mL LIF, 2 mM
Alkaline phosphatase staining and immunofluorescence
Alkaline phosphatase (AP) staining was performed using an AP Detection Kit (Millipore) according to the manufacturer's instructions. For immunofluorescence, cells were fixed with 4% paraformaldehyde for 30 min at room temperature. After washing with PBS three times, cells were treated with PBS containing 0.1% Triton X-100 (PBT) for 30 min and blocked with 5% bovine serum albumin (BSA) in PBT for 1 h at room temperature. Cells were incubated at 4°C overnight with the primary antibodies anti-Nanog and anti-Oct4 (Abcam). After three washes with PBT, cells were incubated with secondary antibodies for 1 h at room temperature. The nuclei of all cells were counterstained with 4′,6-diamidino-2-phenylindole (DAPI).
Spontaneous differentiation
To form embryoid bodies (EBs), iPSCs were trypsinized into single cells and prepared at 3×104 cells/mL in ES medium without LIF. Drops (20 μL) were pipetted onto the lid of a petri dish until the surface was covered. Hanging drops were incubated at 37°C for 3 days. EBs were harvested, cultured in suspension for an additional 2 days, and replated onto 0.1% gelatin-coated dishes. Medium was refreshed every other day. Representative lineage-specific markers were evaluated by semiquantitative RT-PCR on day 10 to determine if spontaneous differentiation had occurred. All primers used for PCR are listed in Table 1.
Teratoma formation
Approximately 1×106 iPSCs in PBS were injected subcutaneously into the dorsal flanks of nonobese diabetic/severe combined immunodeficient (NOD/SCID) mice. Four to five weeks after injection, the morphology of formed tumors was analyzed by paraffin sections. SCs without transfection were injected as a control.
Cell cycle analysis and karyotyping
Cell cycle analysis was performed using a Cell Cycle and Apoptosis Kit (Beyotime, China) following the manufacturer's instructions. Briefly, cells were fixed with precooled 70% ethanol at 4°C for 12 h. After centrifugation at 100×g for 5 min, cells were resuspended in 0.5 mL of Propidium Iodide staining solution and incubated at 37°C for 30 min. DNA contents of the cells were analyzed by flow cytometry on a flow cytometer (FACS Aria III, BD Biosciences, San Jose, CA, USA) at 488 nm.
For karyotyping, cells were treated with 0.5 μg/mL colchicine for 2 h, trypsinized, and pelleted by centrifugation at 300×g for 5 min. Cells were resuspended in 6 mL of prewarmed hypotonic KCl solution, incubated at 37°C for 20 min, and fixed for 10 min at room temperature by addition of 5 mL of fresh fixative solution (acetic acid:methanol, 1:3). Cells were then dropped on to cold slides and air dried. Samples were stained with Giemsa solution, and metaphases were observed and photographed on a microscope (BX51WI-FL, Olympus, Japan).
Real-time RT-PCR analysis
Total RNA was extracted with TRIzol, and reverse transcription was performed using Reverse Transcriptase M-MLV (TakaRa, Japan) and random hexamer primers. PCR was performed with specific primers using GoTaq Master Mix (Promega). Real-time PCR was performed using SYBR Green Mix (Roche) as per the manufacturer's instructions. Data were collected with Applied BioSystems 7500 System. All primers are listed in Table 1. Following a 10-min denaturation at 95°C, the reactions were cycled 40 times with a 15-sec denaturation at 95°C and a 60-sec annealing at 59°C.
Statistical analysis
Each experiment was repeated at least three times. Statistical analyses were performed using SPSS version 17.0, and values were expressed as the means±standard error of the mean (SEM). Data were analyzed using analysis of variance (ANOVA) and Fisher's protected least significant difference (LSD) tests. Differences were considered to be significant at p<0.05.
Results
Identification of SCs
SCs expressed vimentin as detected by ICF (Fig. 1), and mRNA for the SC markers SGP-1, SGP-2, Transferrin (TF), ABP, and AMHII were detected by semiquantitative RT-PCR (Fig. 2A). SCs also expressed low levels of Klf4 as expected, but no expression of the ESC-specific genes Nanog, Oct4, and Sox2 was detected (Fig. 2B).
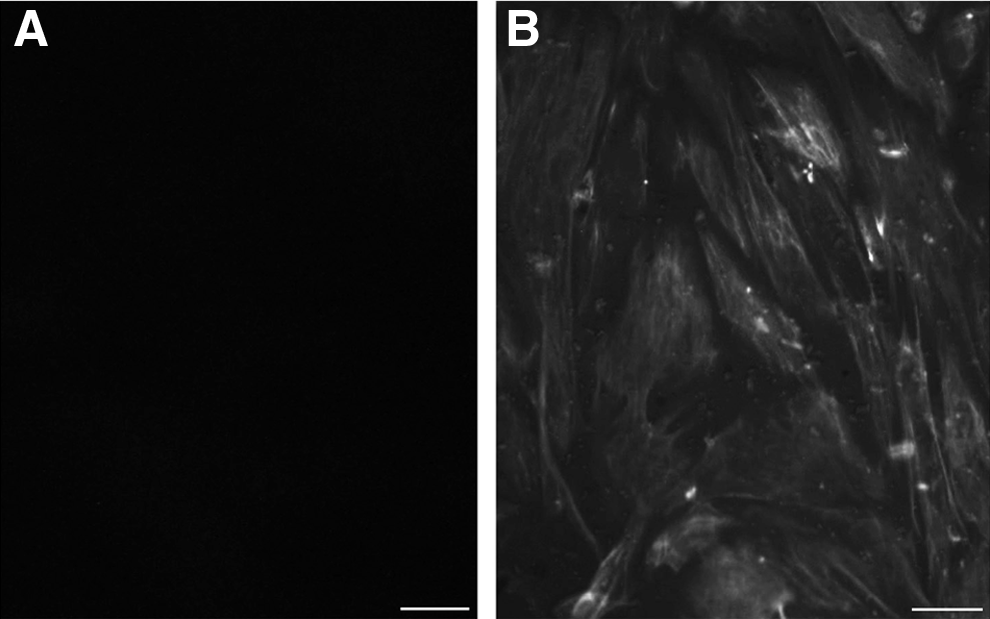
Vimentin, a marker of SCs, was detected by ICF. (
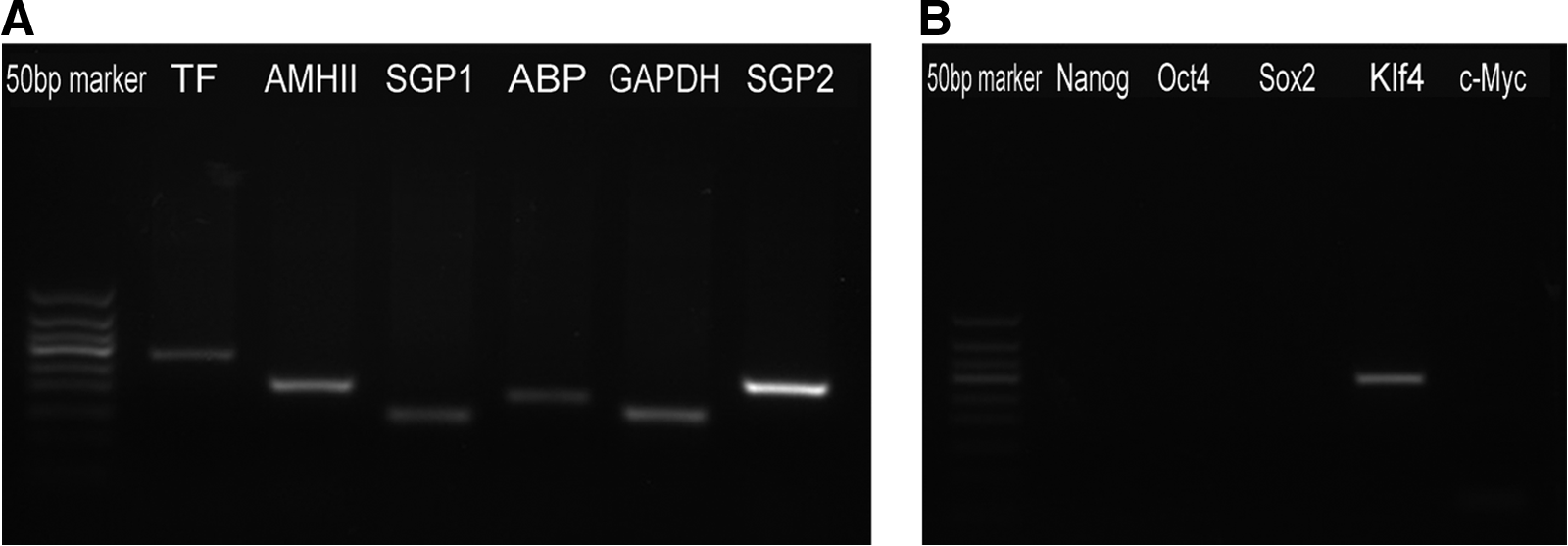
Characterization of isolated SCs. (
Generation of SCiPSCs
The time course for the generation of iPSCs cells from SCs is shown in Figure 3A. Green fluorescent protein (GFP) was introduced to monitor the infection efficiency and to monitor the timing of silencing of the exogenous genes. Because somatic cells are intolerant to KSR, the SC medium was gradually changed to ES serum-free medium to reduce apoptosis. As early as day 3 postinfection, the cells expressed GFP and differed morphologically from uninfected SCs (Fig. 3B). As the culture progressed, the individual cells became smaller and began to form colonies (Fig. 3C). On day 7, compact, round colonies appeared as the colonies continued to grow larger. On the basis of morphology and lack of GFP expression, we picked 20 colonies 18–20 days following transduction. Six of the 20 chosen colonies could be renewed for at least 20 passages, while eight gradually differentiated into other cells. The remaining 14 clones did not survive further passage.
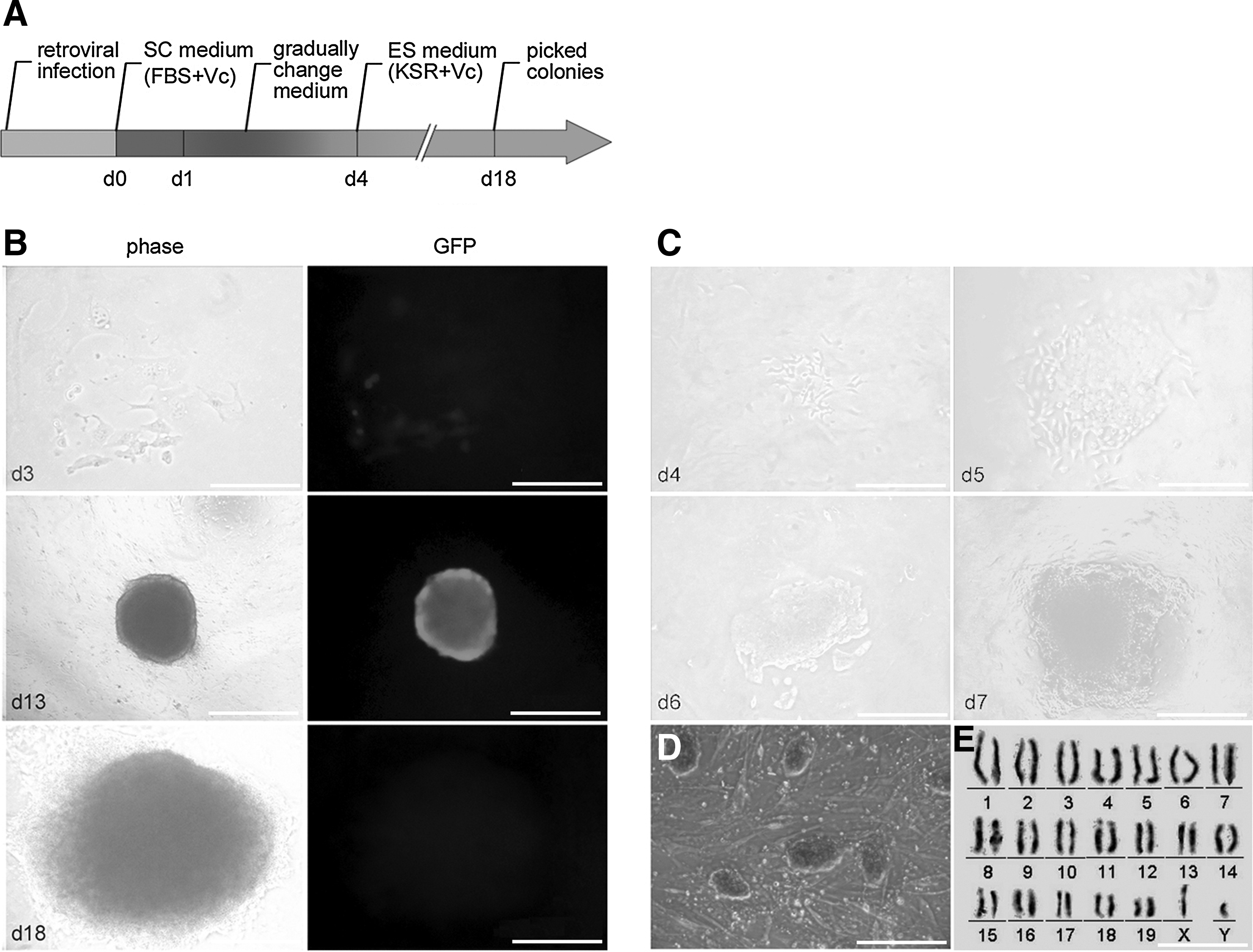
Generation of iPSCs from SCs. (
The morphology of the SCiPSCs was distinguishable from that of R1 mouse ESCs and showed positive AP staining (Fig. 3D). The SCiPSCs maintained a normal karyotype of 40 chromosomes, including X and Y (Fig. 3E).
Characteristics of SCiPSCs
We evaluated the proliferative properties of SCiPSCs (Fig. 4). As shown in Table 2, the three phases of the cell cycle of SCiPSCs (G1, S, and G2/M) were similar to those of ESCs (p>0.05), whereas the cell cycle distribution of SCiPSCs was fundamentally different from that of MEFs or SCs (p<0.05). The proliferative capacity of SCs was significantly lower than that of MEFs (p<0.05).
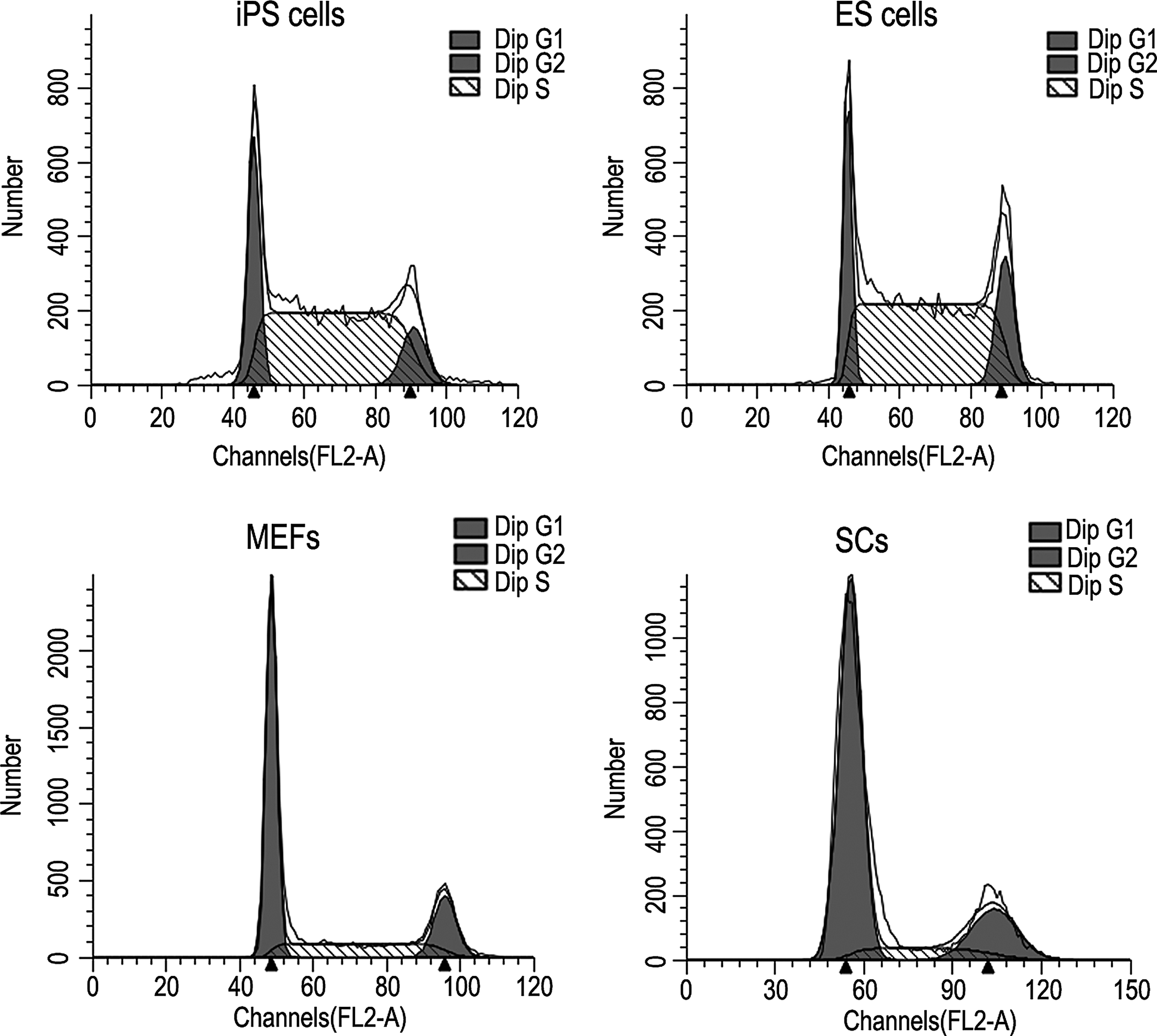
The analysis of the cell cycle distribution of SCiPSCs, ESCs, MEFs, and SCs. Dip, diploid.
Numbers represent means±standard error of the mean (SEM).
Within the same column, values with different superscript letters (a, b, and c) were significantly different (p<0.05).
Dip(diploid); SCiPSCs, iPSCs induced from SCs; ESCs, embryonic stem cells; MEFs, mouse embryonic fibroblasts; SCs, Sertoli cells.
We also investigated the pluripotency of SCiPSCs. Semiquantitative RT-PCR demonstrated that SCiPSCs expressed the majority of ESC marker genes, including Nanog, Oct4, Sox2, Rex1, Fbx15, E-cad, and Fgf4 (Fig. 5A). We selected Oct4 and Fgf4 for quantitative real-time RT-PCR analysis and found that their levels, although lower, were not significantly different from those in ESCs (p>0.05; Fig. 5B). The SCiPSCs were positive for Nanog and Oct4 (Fig. 5C) as analyzed by ICF. Although the transduced cells on day 13 of the reprogramming process did express the four transduced genes (Fig. 5D), they were not expressed in SCiPSCs (Fig. 5E). However, the endogenous counterparts of the four pMXs transgenes were reactivated.
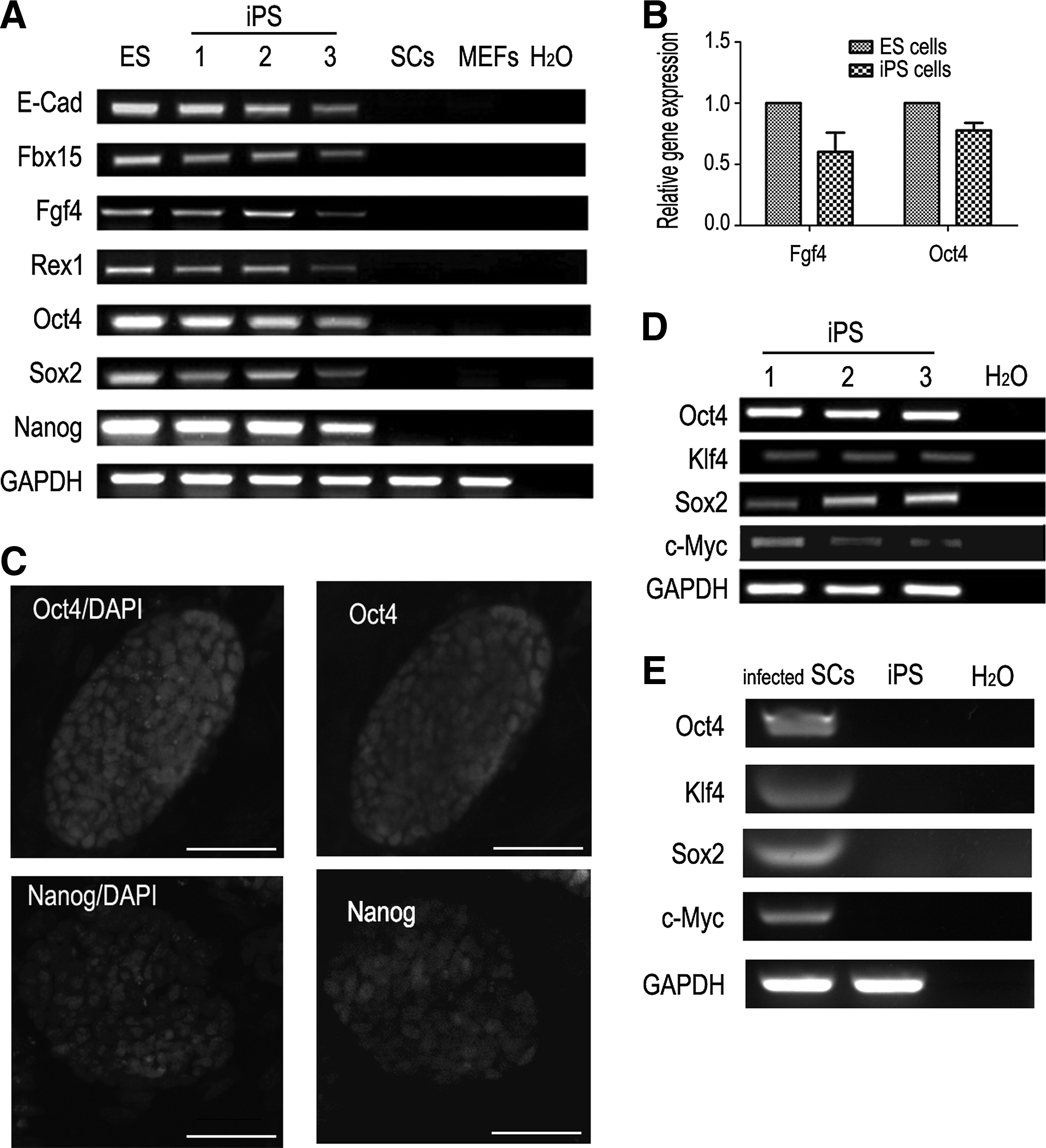
Gene expression pattern of SCiPSCs. (
To confirm the differentiation potential of these SCiPSCs, EBs (in vitro) and teratomas (in vivo) were formed. EBs were obtained using hanging drops and differentiation in gelatin-coated cell dishes (Fig. 6A). SCs injected in the control did not form teratomas or EBs in the drops. Semiquantitative RT-PCR analysis showed that the cells expressed markers of all three primary germ layers, including endoderm derivations (AFP, GATA4, and GATA6), mesoderm derivations (Brachyury), and ectoderm derivations (Nestin, Otx2, Pax6, and βIII-Tubulin) (Fig. 6B). Interestingly, spontaneously contracting patches of cells were also observed. For in vivo differentiation, SCiPSCs were subcutaneously transplanted into NOD/SCID mice. Four to five weeks later, the cell lines gave rise to tumors containing a wide variety of tissues derived from all three germ layers—epidermis and neural tissue (ectoderm), cartilage, adipose, and muscle (mesoderm), and gut-like epithelium (endoderm; Fig. 6C).
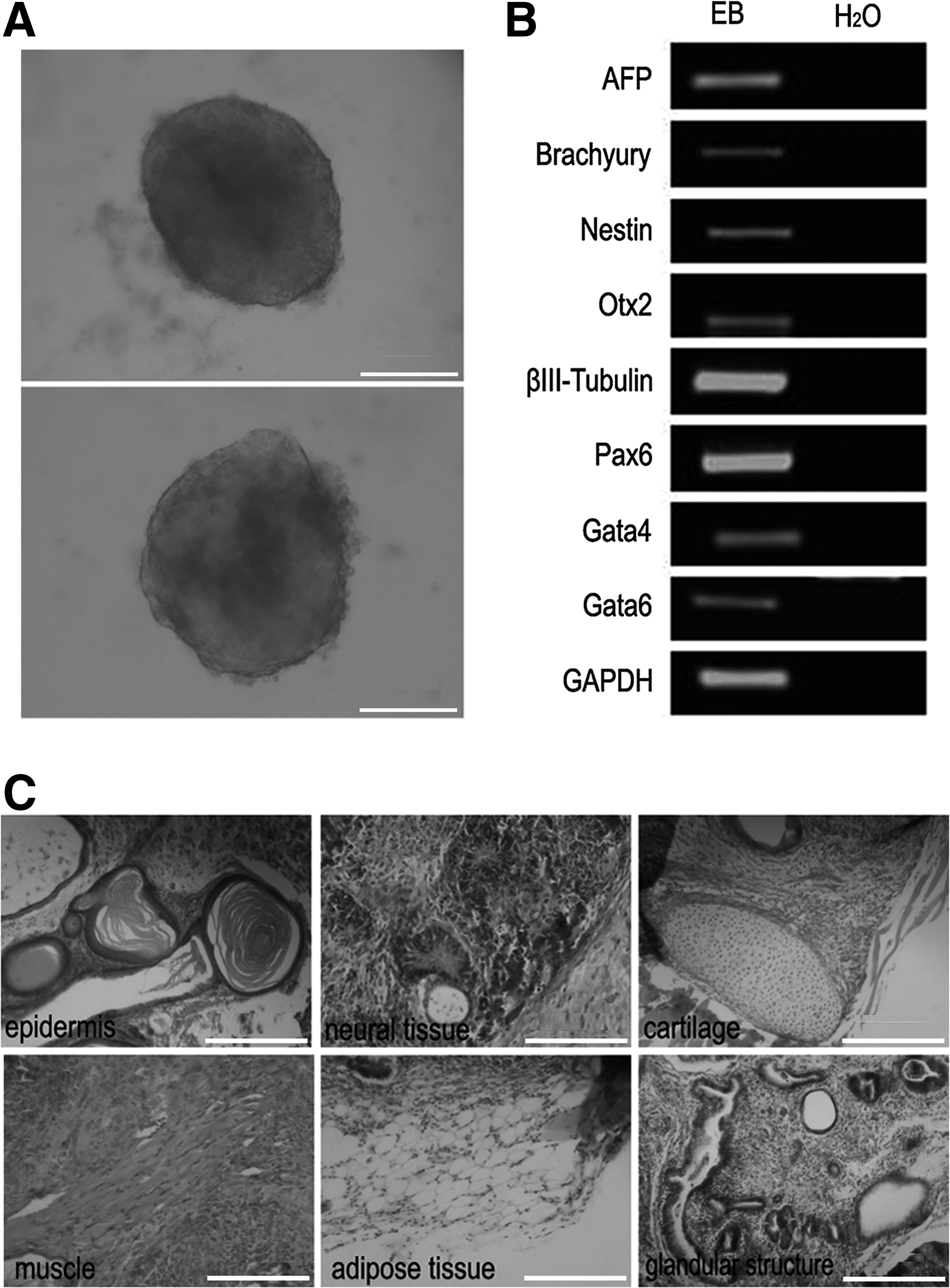
The differentiation potential of SCiPSCs. (
Impact of NO on pluripotency and apoptosis in SCiPSCs
To determine the effects of NO on pluripotency of the SCiPSCs, the expression of the iPSC-specific markers Nanog, Oct4, and Sox2 was analyzed by real-time RT-PCR (Fig. 7A). Expression of Nanog increased at 5 μM DETA/NO (p<0.05) in the absence of LIF, whereas Oct4 and Sox2 levels were maintained. The expression of Sox2 decreased when cells were incubated with 400 μM of the NO inhibitor
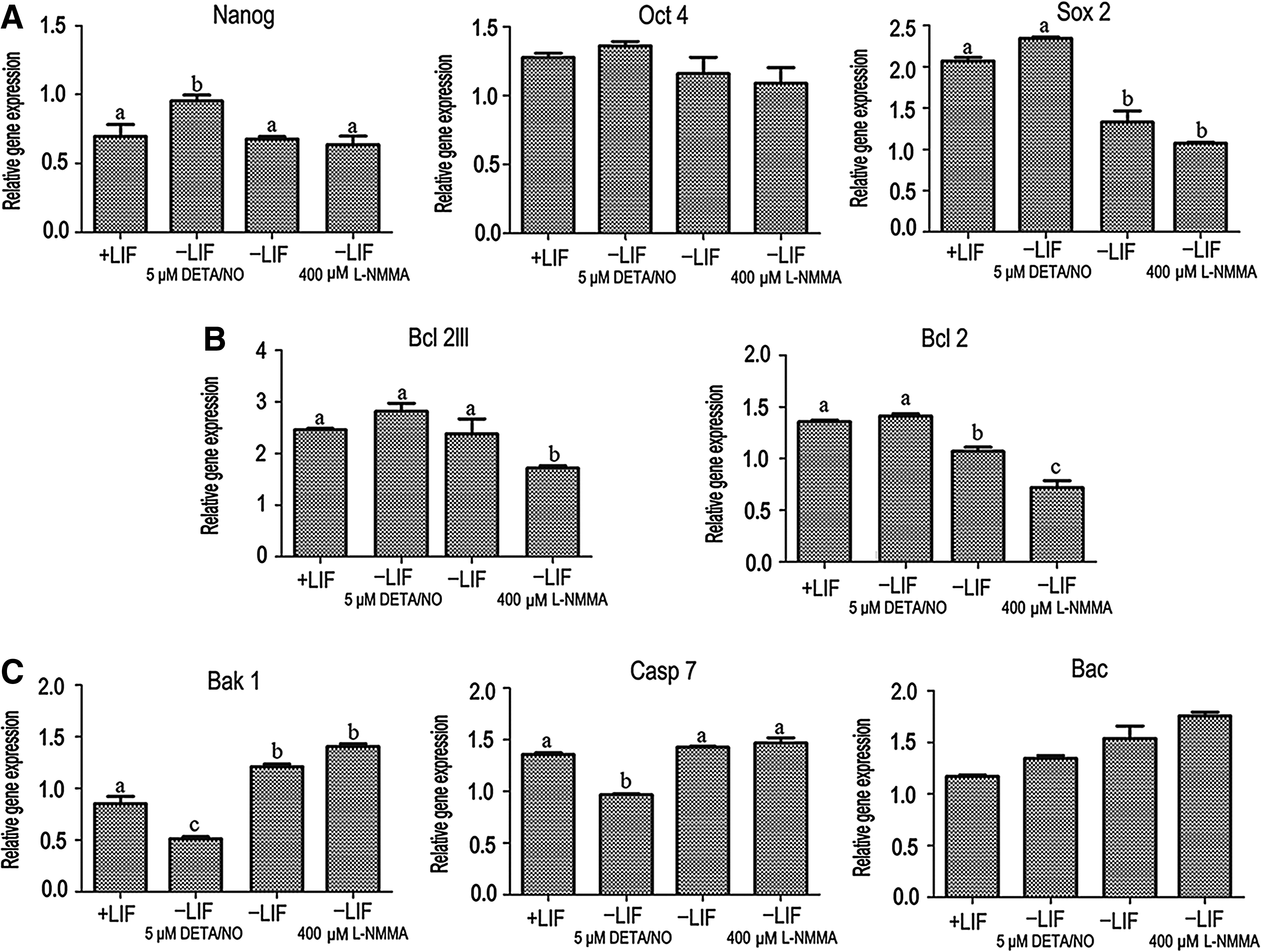
The impact of low NO on gene expression in SCiPS cells. (
We also evaluated the functions of NO on cell survival and apoptosis. DETA/NO (5 μM) maintained expression of Bcl2lll and Bcl2 in LIF-deprived medium (Fig. 7B). The expression levels of the antiapoptotic genes Bcl2lll and Bcl2 decreased at 400 μM
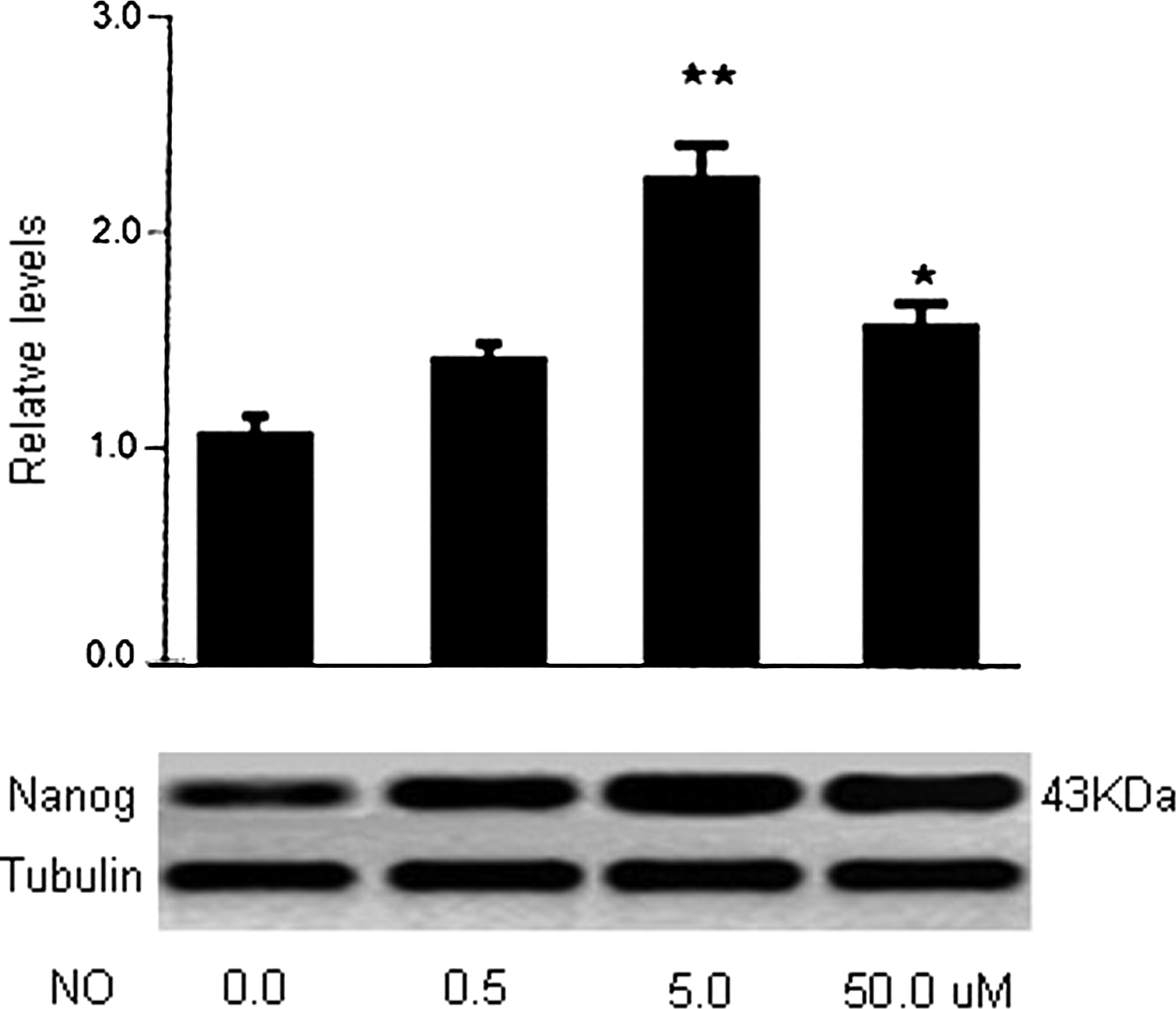
The reaction of Nanog to NETA/NO. In the culture, iPSCs express Nanog in response to NETA/NO in a dose-dependent manner with the maximal expression at 5 μM, as displayed in the figure from western blotting.
Discussion
This study demonstrated that mouse SCs can be reprogrammed by defined factors, resulting in SCiPSCs that express markers similar to those of ESCs and sustain self-renewal and differentiation potential. Reprogramming is not limited to mesoderm derivatives such as SCs: Other types of cells can also be induced into iPSCs, including liver cells and pancreatic β-cells (Aoi et al., 2008; Stadtfeld et al., 2008) from the endoderm, and neural stem cells (Kim et al., 2008; Kim et al., 2009; Shi et al., 2008), dermal fibroblasts (Lowry et al., 2008), and keratinocytes (Aasen et al., 2008) from the ectoderm. In addition, different combinations of factors could induce somatic cells into iPSCs. Several investigations recently demonstrated that defined factors could directly reprogram mature differentiated cells without iPSC intermediates (Caiazzo et al., 2011; Huang et al., 2011; Ieda et al., 2010). Taken together with our results, this suggests that any specific somatic cell could be directly converted into any other type of cells with the appropriate reprogramming factors.
Some studies have reported that infected cells need to be passaged onto feeder layers during the reprogramming progress (Huangfu et al., 2008). However, we found that feeder cells were not essential and that the morphology and characteristics of iPSCs under feeder-free conditions were not significantly different from those on feeder layers. Once the colonies are picked, the SCiPSCs do need feeder cells to prevent differentiation. These data provide evidence that the ectopic expression of the four factors is enough to activate the endogenous gene network in serum-free medium.
The Moloney murine leukemia virus (Mo-MLV)–derived vector pMX was used in the induction of SCiPSCs. The replication of Mo-MLV is restricted to early mouse embryos, mouse embryonic carcinoma (EC) cells, and ESCs (Jahner et al. 1982; Teich et al. 1977). The silencing at the transcription level is largely attributed to retroviral silencer elements and epigenetic regulation (Hotta and Ellis, 2008; Wolf and Goff, 2007). The silencing of retroviral vectors indicates the full reprogramming of iPSCs (Nakagawa et al., 2008; Takahashi et al., 2007a). In this study, GFP was silenced and the four reprogramming factors were inactivated in the selected colonies, suggesting that the silencing of GFP serves as a good marker for the quality of SCiPSCs and that our SCiPSCs were fully reprogrammed.
ESCs are fundamentally different from somatic cells in their cell cycle regulation. The retinoblastoma pathway and the p53-mediated checkpoint control in normal somatic cells were not active in ESCs (Savatier et al., 2002), therefore ESCs have a short G1 phase. Similar results were observed in this study: Most SCiPSCs aggregated in the S phase, suggesting that the cell cycle distribution may serve as a good indicator of iPSCs. The significant difference between MEFs and SCs is likely due to the limited proliferative activity of SCs (Vergouwen et al., 1991).
The presence of LIF was essential for the generation of SCiPSCs. In the absence of LIF, cells formed flat colonies and did not aggregate into round, sharp-edged colonies and lost the characteristics of self-renewal and pluripotency. LIF/STAT3 signaling is sufficient to maintain self-renewal and the undifferentiated state of mouse ESCs (Matsuda et al., 1999; Niwa et al., 1998; Raz et al., 1999), whereas NO signaling is independent of the LIF/STAT3 pathway. NO regulates the expression of c-Src and Akt in ESCs, which is essential for self-renewal of mouse ESCs (Anneren et al., 2004). NO may also prevent apoptosis through the activation of protective genes, such as the Bcl2 protein family in ESCs (Chung et al., 2001; Yamane et al., 2005). Combined with our results, this suggests that NO may be used for culture of iPSCs because of its capacity to regulate pluripotency and self-renewal. However, the regulatory mechanisms of NO on iPSCs require further investigation.
In conclusion, we successfully established iPSCs lines from mouse SCs and found that NO promoted self-renewal and proliferation of the iPSCs. Our results will accelerate the applications of the SC-derived iPSCs to clinics and in making transgenic animals.
Footnotes
Acknowledgments
The authors thank J. Yang for her assistance in IFC, J. Ran and J. Du for their help with cell culture, and Y.A. Cao for her assistance in Western Blot and helpful proofreading. This study was supported by the China National Special Major Project on Transgenic Crops and Farm Animals (project nos. 2011ZX08008-003 and 2013/2014ZX08008-003).
Author Disclosure Statement
The authors declare that there are no conflicts of interest.