
Abstract
Abstract
It is well known that embryos cultured in a group can create a microenvironment through secretion of autocrine and paracrine factors that can support and improve the embryos' development when compared to the embryos cultured individually. In this study, we used a co-culture system for paracrine communication between different kinds of embryos. The results showed that co-culture of porcine parthenogenetic (PA) embryos significantly improved the in vitro development of cloned (nuclear transfer, NT) embryos. To reveal the possible mechanism of communication between the two groups, we isolated exosomes/microvesicles (EXs/MVs) from the PA embryos conditioned medium (PA-CM) through differential centrifugation and identified them through transmission electron microscope and immunoflourescence against exosomal/membrane marker CD9. Furthermore, these EXs/MVs were found to contain mRNA of pluripotency genes (Oct4, Sox2, Klf4, c-Myc, and Nanog), and the PKH67-labeled EXs/MVs could be internalized by the NT embryos. The current study demonstrates that cloned embryos' developmental competence can be improved through co-culturing with PA embryos and revealed, for the first time, that in vitro–produced embryos can secrete EXs/MVs as a possible communication tool within their microenvironment. Moreover, it provides a new paradigm for embryo-to-embryo communication in vitro.
Introduction
E
Porcine cloning and nuclear transfer (NT) have potential application in various aspects of bioscience and biotechnology, such as livestock propagation, endangered species preservation, organ xenotransplantation, and disease model generation. However, the use of this technique is limited owing to its low efficiency (Estrada et al., 2007).
An ever-growing number of studies worldwide has helped to substantiate the essential functions of the cell secreted membrane-derived vesicles, particularly exosomes (EXs) and microvesicles (MVs), and provided new dimensions for the concept of intercellular signaling. These vesicles contain not only proteins but also messenger RNAs (mRNAs) and micro-RNAs (miRNAs) (Lee et al., 2012; Thery, 2011; Valadi et al., 2007), and thereby affect cellular activity via the already made proteins and miRNA or by translation of the transferred mRNAs. Valadi et al. (2007) proposed the name “exosomal shuttle RNA” (esRNA) for those transferred RNAs. These cell membrane-derived vesicles are involved in cell adhesion and signal transfer and provide an important method for cell communication (van der Pol et al., 2012). Evidence of secretion of EXs/MVs has been reported in most cell types, including embryonic stem cells (ESCs) and in vitro–produced embryos (Gardiner et al., 2013; Katsman et al., 2012; Ratajczak et al., 2006).
Several studies have reported embryo co-culture with somatic cells (Desai and Goldfarb, 1998; Funston et al., 1997; Kobayashi et al., 1992), oviduct cells (Bavister, 1988; Lee et al., 2003; Lee et al., 2001; Liu et al., 1998; Mermillod et al., 1993; Van Langendonckt et al., 1996; Xu et al., 2001), and CM (Li et al., 2004a; Li et al., 2004b) to mimic the microenvironment conditions associated with the maternal tract. However, only a single report has studied the co-culture of different kinds of embryos together (Terashita et al., 2011) because of the difficulty in discrimination or separation of different kinds of embryos during the co-culture.
This study was carried out to investigate the effect of co-culture of porcine PA embryos on development of nuclear transfer embryos and whether embryos secrete EXs or MVs as a possible mode of communication between embryos.
Materials and Methods
Chemicals
All chemicals were obtained from Sigma-Aldrich Co. LLC. (St. Louis, MO, USA) unless otherwise stated.
Ovaries and cumulus–oocyte complexes recovery and in vitro maturation
Ovaries were obtained from sows and gilts at a local slaughterhouse and were transported to the laboratory in 0.9% NaCl at 25–30°C. Follicular fluid including cumulus–oocyte complexes (COCs) were aspirated from antral follicles (3–6 mm in diameter) and washed three times with tissue culture medium-199 (TCM-199)-HEPES (Invitrogen, Carlsbad, CA, USA) and selected for in vitro maturation on the basis of morphological features, i.e., a compact multilayered cumulus mass and a dark, evenly granulated cytoplasm. The COCs were cultured in four-well dishes (50 COCs per well; Falcon, Becton Dickinson Ltd, Plymouth, UK) in basic maturation medium, TCM-199 supplemented with 10 ng mL−1 epidermal growth factor (EGF), 0.57 mM cysteine, 0.91 mM sodium pyruvate, 5 μg mL−1 insulin, 1 μg mL−1 follicle-stimulating hormone (FSH) (Antrin, Teikoku, Japan), and 1% (vol/vol) penicillin-streptomycin (Pen-Strep; Invitrogen) at 39°C in a humidified atmosphere of 5% CO2 for 44 h (two stages, with hormonal removal in the second stage). After 44 h, oocytes and expanded cumulus cells were separated by pipetting with 0.1% hyaluronidase in Tyrode's albumin lactate pyruvate (TALP). Denuded oocytes were examined by microscope as free of any attached somatic cells and then were subjected to parthenogenesis or used for nuclear transfer.
Parthenogenetic activation and in vitro culture of matured oocytes
Denuded oocytes were equilibrated sequentially in a gradient concentration (0%, 33%, 66%, and 100%) of mannitol solution (0.25 M) in a four-well dish. The oocytes were then transferred into a two-electrode mannitol chamber connected with a BTX Electrocell Manipulator ECM 2001 (BTX, Inc., San Diego, CA, USA), activated by a single pulse of 1.5 kV/cm for 100 μsec (Okada et al., 2006), and kept for 3 min in the activation medium (Kwon et al., 2011). Electrically activated embryos were equilibrated in reverse order to preactivation to decrease the stress on the oocytes. Activated oocytes were then washed in TALP and transferred into 30-μL microdrops of 4 mM 6-dimethylaminopurine (6-DMAP) covered with mineral oil and cultured for 90 min in an atmosphere of 39°C, 5% CO2, 5% O2, and 90% N2. Presumptive zygotes were washed and randomly distributed to the designed experiment for in vitro culture. Culture medium was serum-free chemically defined porcine zygote medium-5 (PZM-5) (Funakoshi Co., Tokyo, Japan) (Suzuki et al., 2004) covered with mineral oil and cultured in an atmosphere of 39°C, 5% CO2, 5% O2, and 90% N2. On day 2, embryos were evaluated for cleavage to the two-cell stage or beyond. Blastocyst formation was assessed on day 7.
Nuclear transfer
NT was carried out in accordance with our previous protocol (Koo et al., 2009). In brief, in vitro–matured oocytes were enucleated using an aspiration pipette 18 μm in diameter, microinjected with Sinclair male kidney cells, as donor cells, and fused by electrical stimulation. The constructed embryos were then activated, cultured, and checked for development following the same method for PA embryos.
Co-culture of embryos and CM supplementation
PA embryos were co-cultured with cloned (NT) embryos using 24-well plates supported with 0.4-μm Transwell (TW) polyester membrane inserts (Corning Inc., Pittston, PA, USA) to permit embryos communication with a distance of 2 mm between them (Fig. 1) at 39°C in a humidified atmosphere of 5% CO2, 5% O2, and 90% N2. Additionally, we studied the temporal effect of PA embryos CM (PA-CM) supplementation on the development of the same number of cloned embryos. After removing the culture medium, PA-CM of days 2, 4, and 6 was supplemented to the cloned embryos every 2 days using a glass micropipette. The supplementation was either synchronized to the cloned embryos' developmental course (i.e., days 2, 4, and 6 PA-CM were added to the days 2, 4, and 6 cloned embryos, respectively) or preceded by 2 days (i.e., days 2, 4, and 6 PA-CM were added to days 0, 2, and 4 cloned embryos, respectively). Plain PZM-5 was added to the control group in the same manner. The embryo/medium density was one embryo/10 μL in all groups.
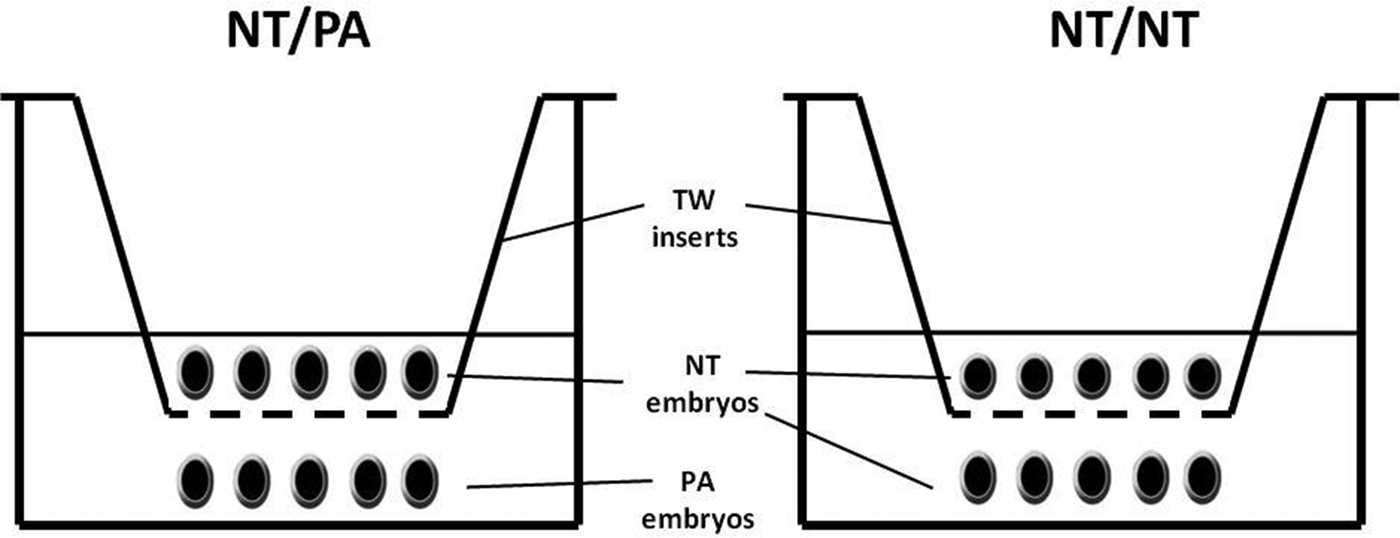
Embryo co-culture design. Cloned embryos (NT) were kept in 0.4-μm Transwell (TW) Polyester Membrane Inserts with a distance of 2 mm from the PA embryos. The medium covers both groups with a density of one embryo/10 μL of PZM-5 medium.
Isolation of EXs/MVs
PA embryos were cultured in groups (one embryo/1-μL PZM-5 microdrops) covered with mineral oil. PA-CM was collected daily via glass micropipettes until the blastocysts hatched. A total of 750 embryos (six replicates of 125 embryos) were used, and the collected CM were kept in −20°C until use. PA-CM of embryos on days 2, 4, 6, and 7 was subjected to differential centrifugation according to the previous protocols (Lässer et al., 2012; Théry et al., 2006; Witwer et al., 2013). In brief, PA-CM was centrifuged at 300×g for 10 min; the supernatant was centrifuged at 2,000×g for 10 min to remove residual cells and debris and at 10,000×g for 30 min to remove apoptotic bodies and cell debris. An equal amount of phosphate-buffered saline (PBS) was added to the supernatant and centrifuged at 200,000×g for 2 h (OptimaTM TLX ultracentrifuge, Beckman Coulter Inc., Fullerton, CA, USA) in 1-mL thick-wall polycarbonate tubes (Beckman Coulter Inc., item no. 343778) to pellet the EXs/MVs. The pellet, which was very tiny, was finally resuspended in 30 μL of PBS. Any EXs would be contained in these pellets, along with MVs. The resuspended pellets were used for checking the presence of MVs/EXs through the electron microscopy, immunofluorescence, RNA extraction, and EXs/MVs labeling. Porcine follicular fluid (PFF) was used as a control positive (da Silveira et al., 2011); the negative control was the plain PZM-5. The same procedures were applied to punctured PA embryos (PPA-CM) (n=750, six replicates of 125 embryos); a hole was made in the zona pellucida using an 18-μm-diameter needle.
Identification of EXs/MVs through transmission electron microscopy and immunofluorescence
To identify the contents of PA-CM and PPA-CM, the resuspended pellets were examined using transmission electron microscopy (TEM) (Ismail et al., 2012). Briefly, 7.5 μL of the pellet suspension was top loaded on 300-mesh grids and dried. The grids were stained in 2% uranyl acetate and visualized with energy-filtering TEM (Carl Zeiss Microscopy GmbH, Oberkochen, Germany) at 120 kV. The mean diameter of the EXs/ MVs was measured in eight microscopy field images (total 40) using ImageJ 1.47t software (National Institutes of Health, Bethesda, MD, USA). For immunofluorescence (IF), we followed the published protocols (Ng et al., 2013; Théry et al., 2006). In brief, 7.5 μL or 0.5 μg of purified pellet protein (measured by NanoDrop 2000 Spectrophotometer, Thermo Fisher Scientific, Wilmington, DE, USA, according to Desjardins et al., 2009) was incubated with 5 μL of 4-μm aldehyde/sulfate latex beads 4% wt/vol (Life Technologies Corp., Grand Island, NY, USA) in a 30-μL final volume of PBS at room temperature for 15 min. PBS (170 μL) was added, and the mixture was incubated in a test tube rotator for 2.5 h at room temperature. Next 22 μL of 1 M glycine/PBS (i.e., 100 mM final concentration) was added and mixed gently (to block the unbound sites of the latex beads) and then let stand on the bench for 30 min at room temperature. The beads were pelleted by centrifugation at 1,500×g for 3 min at room temperature, washed twice with 1 mL of PBS/0.5% bovine serum albumin (BSA). The EX–bead complex incubated with anti-CD9, immunoglobulin G (IgG) conjugated to fluorescein isothiocyanate isomer 1 (FITC; Abcam, catalog no. ab34162, Dawinbio, Seoul, Korea) for 1 h at room temperature. A negative control antibody reaction was performed using normal mouse IgG. The labeled EX–bead complexes were again pelleted and washed twice as above and finally resuspended in 20 μL of PBS/0.5% BSA. Ten microliters of the final complexes were spread on a microscope slide with a coverslip using Vectashield HardSet Mounting Medium (H-1400, Vector Laboratories Inc., Burlingame, CA, USA), air-dried, cover-slipped, and sealed with nail polish. The slides were examined using a HAL 100 fluorescence microscope (Carl Zeiss Microscopy GmbH). Another 10 μL of complexes were resuspended in 150 μL PBS/0.5% BSA for fluorescence-activated cell sorting (FACS) analysis (analyzed by Cell Lab Quanta SC, Beckman Coulter Inc., Fullerton, CA, USA) to estimate the fluorescence signals of the EX–bead complexes. The control positive was the porcine follicular fluid EXs (da Silveira et al., 2011), and the control negative was the PZM-5 in the above-mentioned procedures.
EXs/MVs labeling and internalization by the embryos
EXs/MVs pellets were subjected to fluorescent labeling using PKH67 dye (Sigma-Aldrich), a green fluorescent dye that labels the lipid membranes, according to the manufacturer's instructions and da Silveira et al. (2011), with some modifications. Briefly, a 5-μL EX suspension in PBS was resuspended in 120 μL of diluent C (provided by the manufacturer), mixed with freshly prepared PKH67 in 125 μL of diluent C to reach a final concentration of 5×10−6 M, and incubated for 5 min. Labeling was stopped by addition of an equal volume of 1% BSA in PBS and incubation for 1 min. The labeled EXs/MVs were washed two times in PZM-5 using ultracentrifugation and resuspended in 25 μL of PZM-5. Embryos were incubated with the labeled EXs/MVs for 22 h (da Silveira et al., 2011), washed twice with PBS, and stained with 25 μg mL−1 bisbenzamide for 10 min at room temperature. For negative controls, the embryos were cultured in plain PZM-5 for 22 h. The embryos were washed twice with PBS, mounted with a coverslip using Vectashield HardSet Mounting Medium (Vector Laboratories Inc.), and dried for 5 min at room temperature. Cellular uptake of EXs/MVs was observed under a confocal microscope (Zeiss, Oberkochen, Germany).
Total blastocyst cell count
Six hatching blastocysts from each experimental group were washed in PBS, and the nuclei were stained with 25 μg mL−1 bisbenzamide for 1 h at 37°C. Stained blastocysts were mounted on a glass slide in a drop of glycerol, gently flattened with a cover glass, and examined for cell counting with a fluorescence microscope using a 346-nm excitation filter. Digital photographs were also taken for total cell counting using ImageJ 1.42q software.
RNA extraction and cDNA synthesis
Total RNA was extracted and eluted from embryos (five blastocysts on day 6 of IVC) and EX pellets (7.5 μL), using the easy-spin™ (DNA-free) Total RNA Extraction Kit (iNtRON Biotechnology, Inc., KyungGi-Do, Korea) according to the manufacturer's instructions. RNA purity was evaluated using a NanoDrop 2000 spectrophotometer (Thermo Fisher Scientific) by estimating the ratios of absorbance at 230 nm, 260 nm, and 280 nm; values of A260/A280 of ≥2.0 and A260/A230 >2.0 are accepted and used for reverse transcription (RT). RT was carried out at 42°C for 60 min. Individual RT reactions of a total of 20 μL per reaction was performed using amfiRivert II cDNA Synthesis Premix (GenDEPOT, Barker, TX, USA) according to the manufacturer's instructions.
Because of the difficulty in obtaining a high number of NT embryos for EXs isolation, we analyzed the exosomal RNA contents of NT CM instead and compared it to the PA-CM. We cultured 60 embryos (one embryo/1-μL PZM-5 microdrops, three replicates of 20 embryos) of both NT and PA embryos. The CM were collected on days 2 and 4 of embryo culture. For CM total RNA extraction, it was expected that the RNA content would be too small; we followed the protocol of Mestdagh et al. (2008) with some modifications. Ten microliters of CM were collected by glass capillary pipette and heated at 95°C for 5 min to lyse and release the EXs contents (Malik et al., 2013). The entire lysate was used for pulsed RT to increase RT efficiency as follows: 40 cycles of 16°C for 2 min, 42°C for 1 min, and 50°C for 1 sec, followed by a final inactivation at 85°C for 5 min.
PCR and real-time PCR
cDNA (1–2 μL) of PA-CM of embryos on days 2, 4, 6, and 7 were subjected to PCR using a Maxime PCR PreMix kit-i-StarTaq (Intron Biotech., Seoul, Republic of Korea). The PCR amplification was carried out for one cycle of denaturation at 95°C for 5 min and a subsequent 40 cycles with denaturation at 95°C, annealing for 30 sec, extension at 72–C for 45 sec, and a final extension at 72°C for 5 min. Ten microliters of PCR products were fractionated on a 1% agarose gel (iNtRON Biotechnology, Inc., Korea) and stained with RedSafe ™ (iNtRON Biotechnology, Inc.). In all assays, cDNA template negative and reactions without RT resulted in negative amplification. The positive control was a cDNA of day-6 PA blastocysts, and the negative control was PZM-5.
For relative mRNA quantification, real-time PCR (qPCR) was done according to Takara Bio Inc. guidelines. A total 20-μL PCR reaction was made by adding 100 ng of cDNA, 1 μM forward primer, 1 μM reverse primer, 10 μL of SYBR Premix Ex Taq with ROX reference (Takara Bio Inc. Shiga, Japan), and 6 μL of nuclease-free water (Ambion Inc., Austin, TX, USA). The reaction was carried out using Applied Biosystems StepOnePlus™ Real-Time PCR Systems (Applied Biosystems, Forest City, CA, USA). The thermal profile for real-time PCR was 95°C for 10 min, followed by 40 cycles of 95°C for 10 sec and 60°C for 20 sec. Each transcript was relatively quantified in three replicates by calculation using the 2−ΔΔCt method (Livak and Schmittgen, 2001) for comparison of relative mRNA quantification in embryos after normalizing to the housekeeping gene GAPDH. Each sample was repeated three times. Primer sequences, annealing temperatures, and approximate sizes of the amplified fragments are listed in Table 1. The expressed PCR products were gel purified (QIAquick PCR Purification Kit, QIAGEN, Valencia, CA, USA), and DNA strands were directly sequenced (Macrogen, Seoul, Korea; http://dna.macrogen.com/kor) using the forward primer of the corresponding amplification. The identity of each product (≥95%) was confirmed by sequence homology analysis using the Basic Local Alignment Search Tool (BLAST) at the National Center for Biotechnology Information (NCBI) GenBank (http://blast.ncbi.nlm.nih.gov/).
Statistical analysis
The ratios (cleavage rates and blastocyst formation rates) were evaluated using Pearson's chi-squared test. The means of relative mRNA quantification were compared using Student's t-test. Statistical significance was considered when the p value was less than 0.05.
Results
PA embryos improve cloned embryos in co-culture
In our initial experiment, co-culture of NT embryos with PA (NT/PA) embryos significantly increased cleavage (CR) and blastocyst formation (BL) rates than those cultured with cloned embryos (NT/NT) (Table 2) with the same embryo density (one embryo/10 μL); CR was 80.64% vs. 65.55% (p=0.02) and BL was 21.5% vs. 8.88% (p=0.017). There was no significant difference in day-6 blastocyst total cell count in both groups, p>0.05 (Table 2). Interestingly, within the NT/PA group, the CR in PA subgroup embryos was statistically higher than NT subgroup embryos; 90.9 % vs. 80.64 (p=0.04), whereas there was no difference between the two subgroups in blastocyst formation rate, 31.31% vs. 21.5% (p=0.12), respectively. On the other hand, PA embryos cultured alone (PAO) showed statistically increased CR% and BL% if compared to the cloned embryos in NT/PA group, 91.39% and 39.78% (p=0.006), respectively. Another experiment was performed using PA embryos that were punctured (PPA) with an 18-μm needle and cultured with cloned embryos; they showed no significant difference (p>0.05) in blastocyst formation when NT embryos were co-cultured with intact zona pellucida PA embryos (Table S1) (Supplementary Data are available at www.liebertpub.com/cell/). Additionally, cloned embryos were cultured with the same number of PA embryos (1XPA) or with double the number of PA embryos (2XPA) with maintaining the same embryo/medium density; they showed no significant difference in BL formation (Table S2).
Values in the same row carrying different superscripts are considered statistically significant at p<0.05.
The mean of cell count of six hatching blastocysts±standard deviation (SD).
NT, nuclear transfer (cloned) embryos; PA, parthenogenetic embryos; PAO, parthenogenetic embryos only; TW, activated oocytes placed in the Transwell Inserts; CR, cleavage rate; BL, blastocyst formation rate.
qPCR analysis of cloned embryos
Next, we analyzed the relative mRNA quantification between the NT/PA and NT/NT groups. The mRNA relative expression of each gene in the NT/NT group was arbitrarily set as one-fold. The relative quantitative expression of selected pluripotency genes showed some significant changes between the two groups (Fig. 2A); Oct4 (p=0.003), Klf4 (p=0.01), and Nanog (p=0.001) expression were significantly higher in the NT/PA group, whereas Sox2 (p=0.074) and c-Myc (p=0.11) showed no significant difference when compared with the NT/NT group.
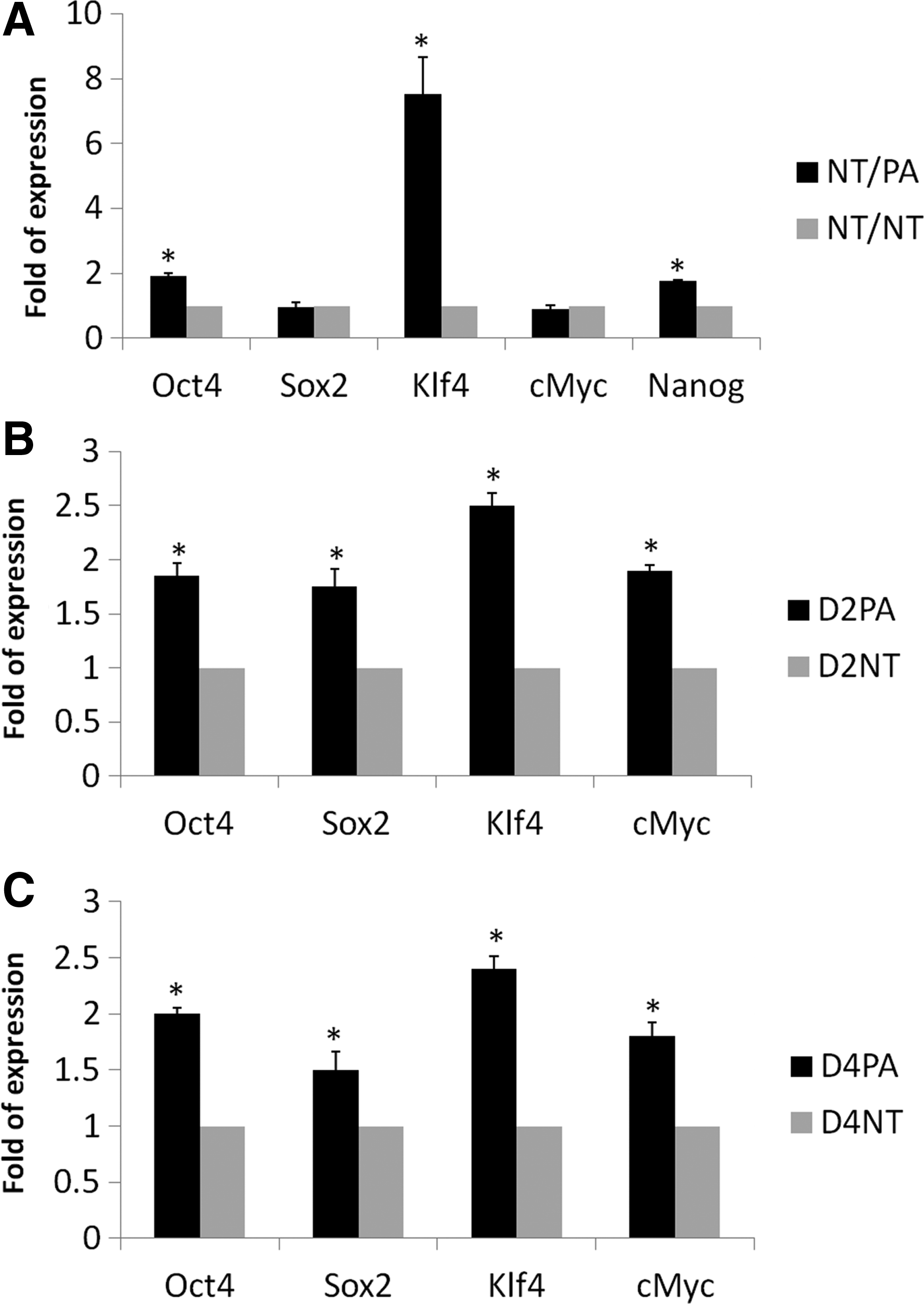
Relative quantitation of pluripotency genes in cloned embryos co-cultured with parthenogenetic embryos (NT/PA) (
Presence of EXs/MVs in the PA-CM
On the basis of these results, we cultured the PA embryos alone to investigate the possible secreted factors that might increase the cloned embryo competence and pluripotency mRNA expression during co-culture. CM was subjected to gradient centrifugation to obtain a pellet. The resuspended pellets were subjected to TEM and IF analysis. In negative-staining TEM, the diameter analysis revealed the presence of varied particles or spheres, ranging from 35.4±6.9 nm (in early embryonic stages, Fig. 3A) to 101.66±18.4 nm, along with the small particles in the late stages of embryo development (Fig. 3B). To characterize these particles, and due to their tiny sizes, we used latex beads to adsorb these particles and immunostained them with fluorescent antibodies against CD9. The results showed that these spheres were CD9+ (Fig. 4A) when compared to the control positive sample, which was the porcine follicular fluid (da Silveira et al., 2011) (Fig. 4B), and control negative sample, which was plain PZM-5 (Fig. 4C). Appropriate gating of FACS analysis demonstrated an intensity of 32.78% in PA-CM against 0.02% in the plain PZM (p<0.01). Experiments were repeated with three independent EXs/MVs preparations with similar results.
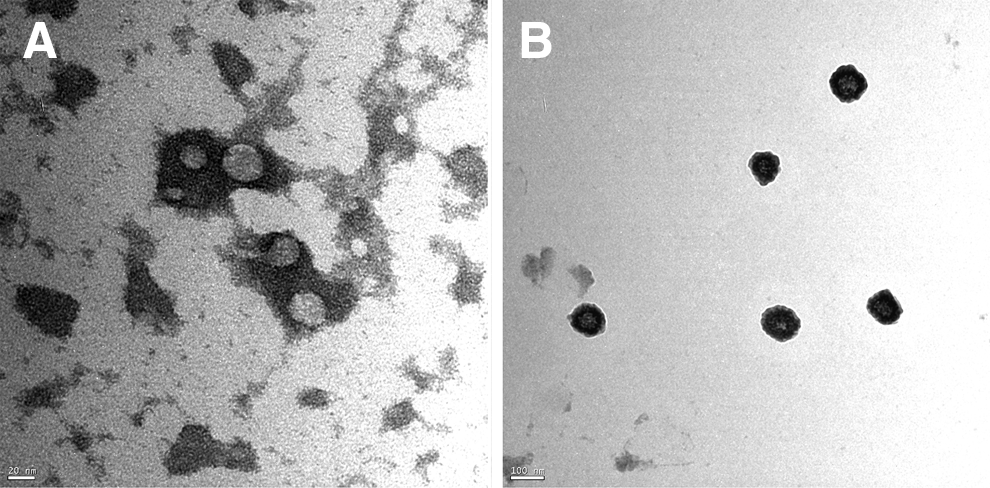
PA embryos–derived EXs/MVs are identified by electron microscope analysis. TEM images show the presence of particles ranged from 30 nm (

PA embryos–derived EXs/MVs are identified by immunofluourescence. Because the EXs size is too small to be analyzed reliably by direct cell sorting, the pellets containing EXs were bound to beads of a size that is in the detection range of the fluorescent microscopes (4-μm diameter latex beads ). The beads were then bound to fluorescent antibodies against CD9 and demonstrated a strong intensity for the exosomal surface proteins CD9 (
Both PA and NT embryos conditioned media contain mRNA of pluripotency genes
Taken into consideration that making an artificial hole in the zona pellucida with an 18-μm diameter needle is sufficient to pass the EXs and MVs, we compared the mRNA contents of pluripotency genes (Oct4, Sox2, Klf4, c-Myc, and Nanog) in the resulting exosomal pellet of intact PA and PPA embryos (Fig. 5). In the early stages (days 2 and 4), there was no significant difference between the expression of all genes except for c-Myc at day-2 embryos which was found in PPA only. In the later stages (days 6 and 7), there was no significant difference between the two groups except in Nanog expression at day 7.
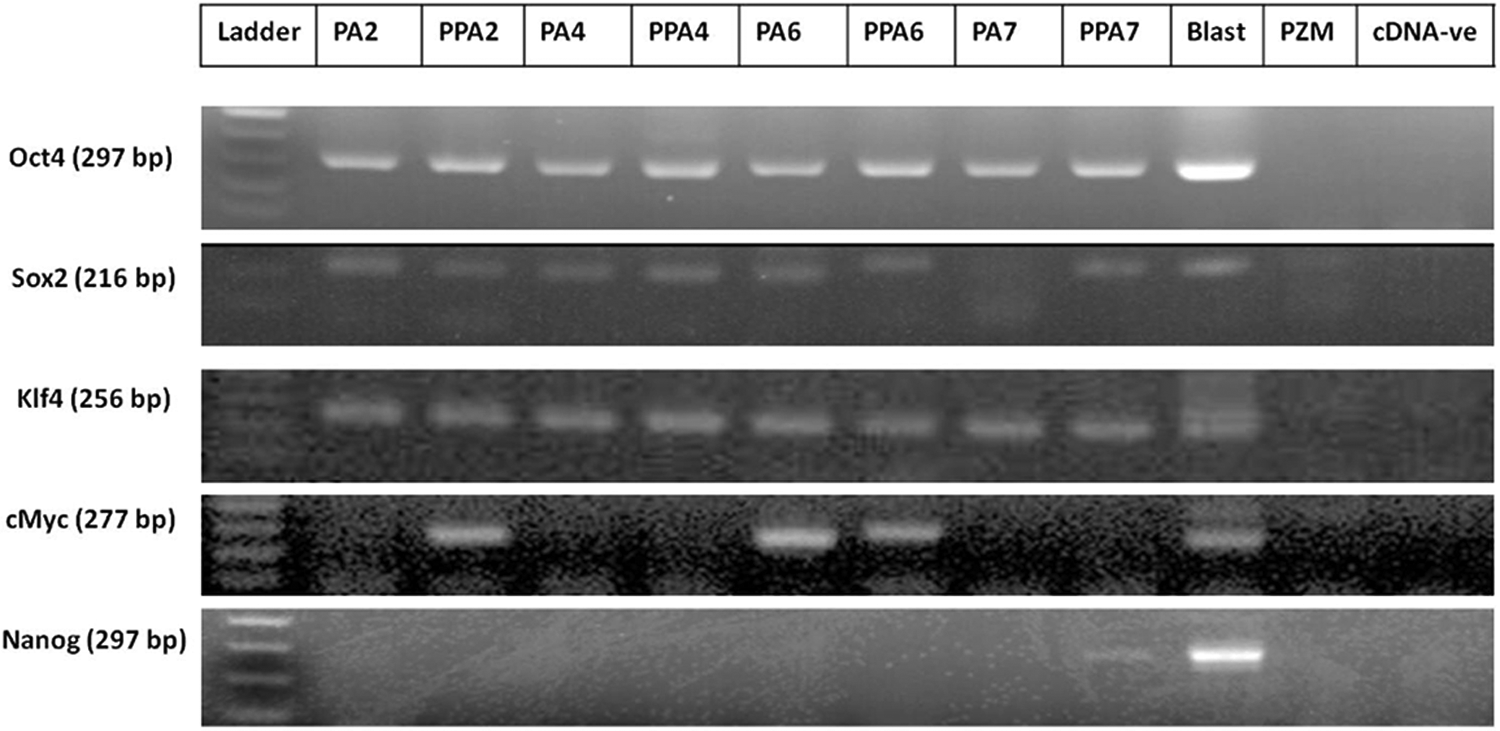
Expression of pluripotency genes in purified EXs/MVs derived from PA embryos. Photomicrograph of gel electrophoresis of PCR products from intact (PA) and punctured (PPA) parthenogenetic embryos on days 2, 4, 6, and 7 of in vitro culture. Day-7 blastocysts (blast) were used as a positive control and plain PZM-5 was used as a negative control (PZM); the other lane was cDNA negative (cDNA-ve) to exclude the primer dimer formation.
Additionally, we compared the relative quantitation of pluripotency mRNAs (Pou5f, Sox2, c-Myc, and Klf4) in CM of small numbers (three replicates of 20 embryos) of PA and NT embryos on days 2 and 4 of embryo culture. The mRNA relative expression of each gene in NT embryos CM was arbitrarily set as one-fold. The relative quantitative expression of selected pluripotency mRNAs in PA embryos was significantly higher than NT embryos either on day 2 or day 4 (Fig. 2B, C).
Labeled EXs/MVs internalization by the developing embryos
To evaluate the possibility of EXs/MVs-mediated transfer of molecules during embryo co-culture, labeled cell membrane EXs/MVs uptake was conducted using the pellets of PA-CM. Purified EXs/MVs were labeled with a green fluorescent PKH67 dye and added to the culture medium of NT embryos. Following a co-incubation period of 22 h, labeled EXs/MVs-treated embryos were washed, fixed, and examined by a confocal microscope. Microscopy results showed effective uptake of PKH67-labeled EXs/MVs by NT embryos (Fig. 6A), whereas NT embryos cultured in plain PZM-5 showed no signals for green fluorescence (Fig. 6B), suggesting that embryo-derived EXs/MVs can be internalized by other embryos sharing the same culture medium.
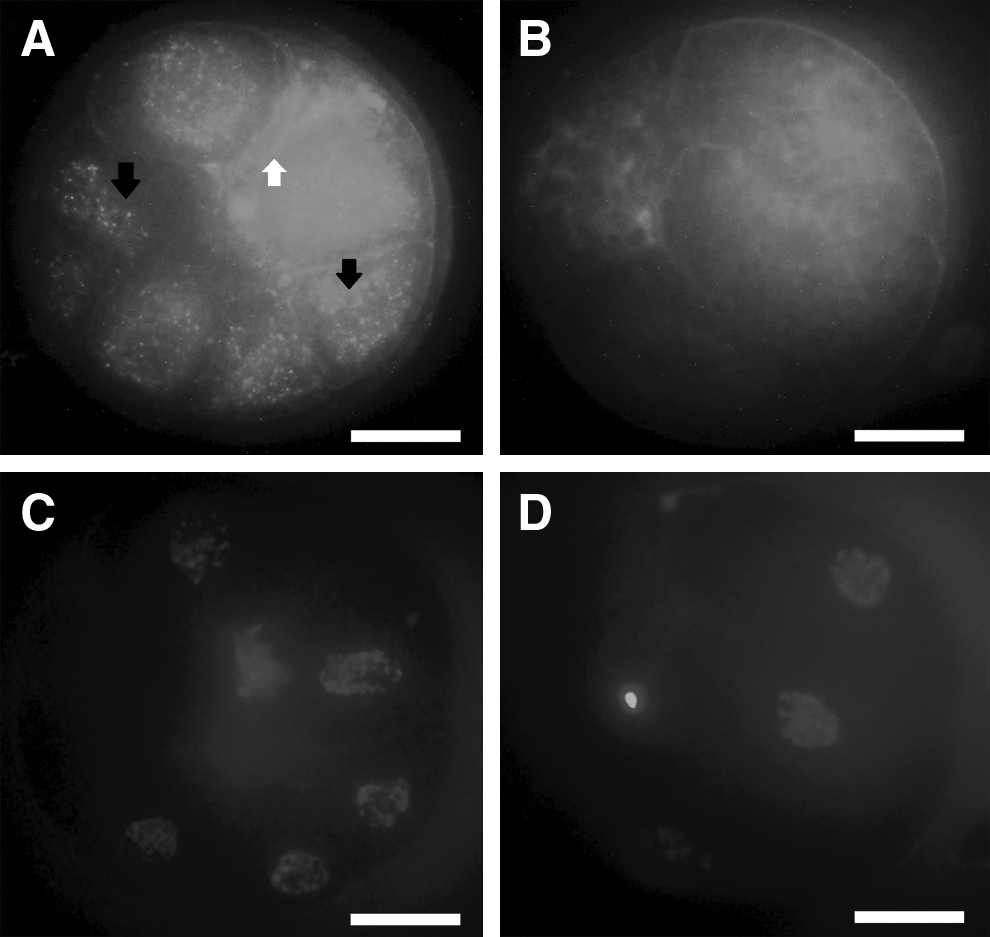
Uptake of PA embryos–derived EXs/MVs by cloned embryos. EXs/MVs purified from PA-CM were labeled with PKH67 dye and added to day-2 cloned embryos for 22 h. PKH67-labeled EXs/MVs emitted fluorescent signals (
PA-CM cannot improve NT embryo developmental competence
PA-CM from days 2, 4, and 6 were supplemented either synchronized to the embryo development course (PA-CM of days 2, 4, and 6 were transferred on days 2, 4, and 6, respectively) or preceded by 2 days (PA-CM of days 2, 4, and 6 were transferred on days 0, 2, and 4, respectively). Supplementation of CM in both groups showed no significant effect on embryo development (Table 3) (p>0.05).
Control group was supplemented by plain porcine zygote medium-5 (PZM-5) on days 2, 4, and 6.
Conditioned medium of cultured parthogenetic (PA) embryos of days 2, 4, and 6 were transferred on days 0, 2, and 4, respectively.
Conditioned medium of cultured PA embryos of days 2, 4, and 6 were transferred on days 2, 4, and 6, respectively.
CM, conditioned medium; CR, cleavage rate; BL, blastocyst formation rate.
Discussion
The early preimplantation mammalian embryo is known to be relatively autonomous and can regulate its own development independently until hatching in vitro (Schultz and Heyner, 1993). Nevertheless, a number of previous reports proved that in vitro culture of embryos is remarkably successful when the embryos are kept in large groups during the whole culture period (Ferry et al., 1994; Hoelker et al., 2008; Paria and Dey, 1990). Embryos can generate their microenvironment through secretion of complex and enigmatic trophic autocrine/paracrine growth factors, termed the “secretome” (Bormann et al., 2006; Hoelker et al., 2010; Katz-Jaffe et al., 2006; O'Neill, 1998; O'Neill, 2008). Particularly, cloned embryos (NT) are more sensitive to culture (Yamanaka et al., 2009b), and because of the labor required, few embryos are produced and cultured in vitro.
Here, we attempted to improve the porcine cloning efficiency through co-culture with PA embryos. We used a new co-culture system for embryos consisting of Transwell Membrane Inserts (0.4-μm pores) to separate large groups of different kinds of embryos (NT and PA) and permitting dynamic paracrine interaction through the exchange of their microenvironments. In our results, cloned embryos that were co-cultured with PA embryos showed a significant increase in developmental competency in terms of embryo cleavage (CR=80.64%) and blastocyst formation (BL=21.5%). The cloned embryos were cultured separately [65.55% and 8.88%, respectively (p<0.05)] (Table 2). This result is consistent with the result of Terashita et al. (2011), who showed that co-culture of PA but not IVF embryos with cloned ones, using the well-of-the-well (WOW) system, increased blastocyst formation in cloned embryos. However, this system is beneficial only for using few embryos in a small amount of culture medium (Vajta et al., 2008).
Interestingly, in the NT/PA group, NT and PA subgroups showed no significant difference in blastocyst formation rate between them (Table 2); however, there was a significant difference between these subgroups and those of cultured PA embryos (PAO; PA/PA) (Table 2).
Moreover, the RQ (relative quantification) of Oct4, Klf4, and Nanog showed a significant increase in co-cultured cloned embryos than that of control ones (Fig. 2A), which might be the reason for increasing co-cultured cloned embryo competency. Oct4, Sox2, c-Myc, Klf4, and Nanog are all considered the pluripotency genes in most mammals, with differences in temporal and spatial expression in different animals (du Puy et al., 2011). These genes are the key players in reprogramming of the NT–derived embryos, and they were reported to be lower in NT embryos if compared to other in vitro–derived embryos (Niwa et al., 2000; Rodríguez et al., 2012; Zhang et al., 2011; Zhou et al., 2013). The full mechanism of network interaction between these genes is still not elucidated in the pig because of their unique expression in this species (du Puy et al., 2011); however, there were some published reports regarding the mutual interaction of two (Rodda et al., 2005) or more pluripotency genes (du Puy et al., 2011). The increase in RQ of some pluripotency genes in cloned embryos in the current study can be explained simply because of sharing the microenvironment or secretome that was generated by the in vitro–competent PA embryos (CR=90.9% and BL%=31.31%; Table 2).
To investigate the possible reason for the increment in these mRNAs in co-cultured embryos, we cultured the PA embryos separately and collected their CM (PA-CM), which was analyzed through TEM after differential centrifugation. Interestingly, small particles of 30–120 nm diameter (Fig. 3A, B) were observed; they were <40 nm in the two-cell stage until blastocyst formation and <120 nm from the blastocyst stage expansion until hatching. To characterize these particles, the pellet contents were adsorbed to latex beads and were subjected to fluorescent antibodies against CD9. The beads were found to be CD9+, which confirmed that these structures are membrane-derived vesicles, EXs, or MVs (Fig. 4A). The tetraspanin CD9, a membrane-bound protein, is commonly used to identify EXs/MVs (Mathivanan and Simpson, 2009) and is involved in cell packaging of proteins in EXs (Chairoungdua et al., 2010). Moreover, it is expressed in the oocytes and required for fertilization (Barraud-Lange et al., 2012; Li et al., 2004c; Miyado et al., 2000) as well as expressed in the blastocysts (Goissis et al., 2009). We confirmed CD9 expression in different stages of PA embryos (Fig. S1).
Next, we analyzed the mRNA of pluriopotency genes in the contents of the isolated pellets by PCR. We compared the mRNA expression in the pellets derived from intact PA and punctured PA embryos, because the artificial hole that was made by an 18-μm needle is sufficient to pass the EXs/MVs. All of pluriopotency genes (Oct4, Sox2, c-Myc, Klf4) were conspicuously expressed except Nanog, which was found to be expressed in the late stage of blastocysts (Fig. 5); this is consistent with previous studies (du Puy et al., 2011). We compared our mRNA results to the Vesiclepedia (http://microvesicles.org), which is a compendium database of extracellular vesicles and exosomal proteins, RNAs, and lipids (Kalra et al., 2012), to validate our findings and found that all target mRNAs in our study have been identified by other groups in EXs/MVs from different cell types (Kalra et al., 2012).
In our results, some mRNAs were found to exist either along with the embryo developmental course (Oct4, Sox2, and Klf4) or intermittently expressed (c-Myc) in the EXs/MVs. From this comparison, we assume the presence of the same structures that are able to pass either from an artificial hole or from the intact zona pellucida and confirmed by the results of the development of cloned embryos that were co-cultured either with intact (PA) or with punctured (PPA) zona pellucida (Table S2); there was no significant difference between the two groups (p>0.05). Therefore, it can be hypothesized that the mRNA was transferred from PA embryos to cloned embryos through the EXs/MVs.
Moreover, we compared the relative quantification of the candidate mRNAs in PA and NT embryo CM. To release the mRNA cargoes from the EXs/MVs, we heated the CM at 95°C to lyse the EXs/MVs. The whole lysate was used for real-time PCR analysis. The relative quantification of Oct4, Sox2, Klf4, and c-Myc was significantly increased in the CM of PA embryos than in those of cloned ones on days 2 and 4 of embryos culture when normalized to the GAPDH mRNA of both groups (Fig. 2B–C).
Following the confirmation of presence of mRNA in the CM and purified EXs/MVs, we examined the uptake of EXs/MVs by cloned embryos. EXs/MVs were labeled with PKH67 fluorescent dye and were co-cultured with cloned embryos in vitro for 22 h. The fluorescent microscopy revealed the presence of green fluorescent signals in cultured cloned embryos (Fig. 6). Importantly, the fluorescent microscopy results also showed that the labeled EXs/MVs were taken up by the blastomeres; there were no fluorescent signals in plasma membrane of the blastomeres (Fig. 6A), indicating that EXs/MVs have been internalized in to the blastomeres.
During the past decade, membrane-derived EXs/MVs have received much attention regarding intercellular communication because compelling experimental evidence has proved their involvement as bioactive organelles in carrying nucleic acid and protein cargoes between the cells (for review, see Shifrin et al., 2013). But can EXs/MVs pass through the intact zona pellucida? To answer this question, we have to look into the porcine zona pellucida porosity and permeability. The zona pellucida appeared by scanning EM to be a sponge with a complex fibrous network interspersed with numerous pores (Van Soom et al., 2010). The physical dimension of the zona pellucida differed from in vivo– and in vitro–derived zygotes (Funahashi et al., 2001). The zona pellucida was found to be more porous in the latter and made in vitro–produced embryos more prone to pathogen entrapment and passage of nanostructures (Kim et al., 1996). This may be because of the handling of oocytes and embryos during denuding and culture that affects the zona pellucida integrity and porosity. Interestingly, the zona reaction and subsequent changes in the zona surface differs between IVF and PA embryos; the zona pellucida resumed its porous characteristics after parthenogenetic activation of bovine oocytes (Suzuki et al., 2000). Molecule permeability through the zona depends on the molecule's size and physicochemical factors, such as hydrophilic–lipophilic properties; lipid-containing molecules in particular penetrate the zona pellucida with relative ease (Turner and Horobin, 1997). In the pig, some viruses can penetrate the zona and reach inside the embryos (Bane et al., 1990; Mateusen et al., 2006). Moreover, fluorescent inert microspheres with sizes of 20 nm and 26 nm were able to cross the zona; however, 200-nm microspheres cannot (Mateusen et al., 2004). There was a wide range between the particles used in this experiment, around a seven-fold difference. Taken together, according to our findings and the previous reports, we can suggest that the pig zona pellucida can permit the passage of inert particles less than 200 nm in diameter. This is consistent with our findings; EXs/MVs were less than 40 nm in early-stage embryos and less than 120 nm in late-stage embryos (Fig. 3A, B).
In the current results, some embryonic mRNAs were increased significantly (Oct4, Klf4, and Nanog; Fig. 2A), and other genes showed no significant difference with the control group (c-Myc and Sox2; Fig. 2A), suggesting a network interaction/feedback mechanism between the endogenous and exogenous genes to reach cellular balance or homeostasis to adjust the cellular activity after transfer of external mRNAs.
Furthermore, we explored the temporal effect of PA-CM supplementation on the development of the cloned embryos. PA-CM was supplemented either along with the embryo development course or preceded by 2 days showed no significant effect on embryo development (Table 3); however, it showed some tendency (p=0.26) to adversely affect embryo development. This paradox suggests a temporal interaction of the transferred mRNAs (du Puy et al., 2011; Kim et al., 2008; Ng and Surani, 2011) and reflects the dramatic changes in the daily output of the embryos secretome (Katz-Jaffe et al., 2006). This result also provides the importance of embryo co-culture conditions and the continuous supply of embryo-derived factors to improve cloning efficiency in porcine embryos.
We suggest that the increment of the pluripotency genes is caused by continuous transfer of mRNA cargoes from PA to NT embryos via EXs/MVs, but the acute transfer through CM supplementation paradoxically affects embryo development. It was suggested that foreign mRNA's stability in cells is often tightly and coordinately regulated and the transcriptional rates are low; these mRNAs are rapidly turned over with half-lives of 20–40 min (Rajagopalan and Malter, 1996; Wisdom and Lee, 1991). Therefore, continuous supplementation of mRNA, which was achieved by co-culture, is advantageous over the acute transfer of mRNA by the CM, confirming the concept of dynamic microenvironment or the niche among the cultured embryos.
In summary, our result showed the usefulness of co-culturing PA embryos with cloned embryos, as indicated by increasing cleavage rate and blastocyst formation in the cloned embryos. We suggest that in vitro–derived embryos can secrete EXs/MVs in their CM as possible new mediators within the embryonic microenvironment and embryo-to-embryo communication.
Footnotes
Acknowledgments
We thank Marek Molas, PhD, for assistance in the statistical analysis. This study was financially supported by grants from IPET (#311011-05-3-SB010), MOTIE (#10033839-2013), Research Institute for Veterinary Science, the BK21 PLUS program, and TS Corporation.
Author Disclosure Statement
The authors declare that no conflicting financial interests exist.
References
Supplementary Material
Please find the following supplemental material available below.
For Open Access articles published under a Creative Commons License, all supplemental material carries the same license as the article it is associated with.
For non-Open Access articles published, all supplemental material carries a non-exclusive license, and permission requests for re-use of supplemental material or any part of supplemental material shall be sent directly to the copyright owner as specified in the copyright notice associated with the article.