
Abstract
Abstract
Induced pluripotent stem cells (iPSCs) share similar characteristics of indefinite in vitro growth with embryonic stem cells (ESCs) and may therefore serve as a useful tool for the targeted genetic modification of farm animals via nuclear transfer (NT). Derivation of stable ESC lines from farm animals has not been possible, therefore, it is important to determine whether iPSCs can be used as substitutes for ESCs in generating genetically modified cloned farm animals. We generated ovine iPSCs by conventional retroviral transduction using the four Yamanaka factors. These cells were basic fibroblast growth factor (bFGF)- and activin A–dependent, showed persistent expression of the transgenes, acquired chromosomal abnormalities, and failed to activate endogenous NANOG. Nonetheless, iPSCs could differentiate into the three somatic germ layers in vitro. Because cloning of farm animals is best achieved with diploid cells (G1/G0), we synchronized the iPSCs in G1 prior to NT. Despite the cell cycle synchronization, preimplantation development of iPSC-NT embryos was lower than with somatic cells (2% vs. 10% blastocysts, p<0.01). Furthermore, analysis of the blastocysts produced demonstrated persistent expression of the transgenes, aberrant expression of endogenous SOX2, and a failure to activate NANOG consistently. In contrast, gene expression in blastocysts produced with the parental fetal fibroblasts was similar to those generated by in vitro fertilization. Taken together, our data suggest that the persistent expression of the exogenous factors and the acquisition of chromosomal abnormalities are incompatible with normal development of NT embryos produced with iPSCs.
Introduction
A
Although iPSCs have been used to produce cloned mice by NT with equivalent success to ESC donors (Kou et al., 2010; Zhou et al., 2010), a large joint effort failed to produce cloned piglets using porcine iPSCs, and successful term development was only obtained when iPSCs were differentiated before using them as NT donors (Fan et al., 2013). It is not clear what prevented postimplantation development of embryos reconstructed with undifferentiated iPSCs, but it is possible that either the lack of cell cycle synchronization prior to NT or the quality of the iPSCs compromised the developmental competence of those embryos. Pluripotent cells (ESCs, iPSCs, and blastomeres) have a shortened overall cell cycle length with an extended S phase. This means that using cycling cells as nuclear donors will result in poor development, because S-phase donor cells are incompatible with normal development after NT (Campbell and Alberio, 2003; Campbell et al., 1996a). Cloned farm animals are commonly produced using donor differentiated cells arrested in G0/G1 of the cell cycle (Campbell et al., 1996b; Kasinathan et al., 2001; Wells et al., 2003; Wilmut et al., 1997). Although some reports show that G2/M donors can also be used (Lai et al., 2001; Lai et al., 2002), other reports have found variable levels of aneuploidy and compromised development of the resulting reconstructed embryos (Alberio et al., 2000; Yuan et al., 2014; Zhang et al., 2004).
It is well accepted that NT with differentiated cells is very inefficient. In contrast, some studies have reported higher cloning efficiencies (live offspring) with mouse ESCs (Eggan et al., 2001; Jaenisch et al., 2002; Rideout et al., 2000; Wakayama et al., 1999). Despite higher development to term, the preimplantation development of ESC-reconstructed embryos is lower (15–29%) than embryos reconstructed with differentiated cells (40–67%) (Wakayama et al., 1998; Wakayama et al., 1999; Wakayama and Yanagimachi, 1999). It is possible that the lower preimplantation development of ESCs is due to lack of cell cycle synchronization between donor cells and recipient cytoplasts prior to NT.
In this study, we evaluated whether cell cycle synchronization into G1 could improve preimplantation development of NT embryos using ovine iPSCs compared to parental fetal fibroblasts (PFFs). Reprogramming of key developmental genes (NANOG, SOX2, CDX2, and GATA3) was analyzed in cloned blastocysts produced with iPSCs and PFFs as well as in control in vitro fertilization (IVF) blastocysts. We found that reprogramming of NANOG and SOX2 was abnormal in iPSC-NT blastocysts compared to IVF and PFF-NT blastocysts. In addition, iPSC-NT blastocysts showed persistent expression of the transgenes. Our data suggest that the quality of ovine iPSCs, rather than cell cycle synchronization, is the crucial factor determining normal embryo development after NT.
Materials and Methods
Generation of ovine iPSCs and culture of cells
The transfection of human embryonic kidney (HEK) cells with individually packaged Oct4 (O), Sox2 (S), Klf4 (K), and c-Myc (M) viral vectors was performed as previously described (Breton et al., 2013). Forty-eight hours after transfection, supernatants (6 mL) were collected and filtered through 0.45-μm filters. Ovine PFFs (at passage four) were seeded the day before onto a gelatin-coated six-well plate at 2×105 cells/well and transduced with 1 mL of each of OSKM virus supplemented with 4 μg/mL Polybrene (Sigma). The HEK cells were fed with 6 mL of fresh medium and the transduction was repeated 24 h later. Forty-eight hours after first transduction, transduced PFFs were passaged using 0.25% trypsin-EDTA and seeded at 2×104 cells/well of a gelatin-coated six-well feeder plate. Two days later, the culture medium was changed to iPSC medium [knockout Dulbecco's Modified Eagle Medium (KO-DMEM; Life Technologies) containing 20% knockout serum replacement (Life Technologies), 2 mM Glutamax (Gibco), 0.1 mM nonessential amino acids (NEAA; Life Technologies), 0.1 mM 2ME (2ME; Life Technologies), and supplemented with 10 ng/mL human basic fibroblast growth factor (bFGF; Peprotech)]. Thereafter, the medium was changed daily.
Transduced cells were passaged after 7 days and plated onto six-well feeder plates at 5×103 cells/well. iPSC colonies were picked as they appeared and transferred to gelatin-coated 24-well feeder plates. iPSCs were split 1:2 every 2–3 days. For passaging the cells, 0.05% trypsin (Gibco) was used. iPSCs were incubated at 39°C, 5% CO2, 5% O2 in a humid incubator. PFFs were cultured in (Sigma) supplemented with 10% fetal bovine serum (FBS), 2 mM Glutamax, 0.1 mM NEAA, and 0.1 mM 2ME and incubated at 39°C, 5% CO2, 5% O2. Mouse embryonic fibroblasts (MEFs) were cultured on 0.1% gelatin-coated flasks and maintained in DMEM supplemented with 10% FBS and 2 mM
In vitro differentiation
To test the ability of the clones to differentiate into the three germ layers (endoderm, ectoderm, and mesoderm), we allowed the cells to form embryoid bodies (EBs) and then differentiate. iPSCs were passaged with collagenase and transferred onto bacterial-grade nonadherent petri dishes without any feeder layer, fed with 2 mL of fresh iPSC medium, and incubated at 37°C and 5% CO2 overnight. The next day, the growing cells were transferred to a centrifuge tube and allowed to settle down by gravity for 20 min. The medium was changed to 6 mL of differentiation medium per tube [80% KO-DMEM, 10% fetal calf serum (FCS), 1×NEAA, 2 mM
Colony formation assay
To assess the self-renewal and proliferation abilities of iPSCs under different treatments, a colony formation assay was carried out. A 24-well plate (Nunc) was treated with 0.1% gelatin. MEFs were seeded at 6×104 cells/well, and iPSCs were then seeded at 2000 cells/well. To block fibroblast growth factor (FGF) receptors (FGFRs), MEK, and ALK5 receptors, the following pharmacological inhibitors (from Tocris Bioscience) were used: PD173074 (100 nM), PD0325901 (400 nM), and SB431542 (20 μM), respectively. The medium was changed every day, and cells were allowed to grow for 10 days. Cells were then stained for alkaline phosphatase (AP) activity with a Leukocyte Alkaline Phosphatase Kit (Sigma) according to the manufacturer's instructions. Pictures were taken under bright-field microscopy.
Immunofluorescence staining
Cells were fixed with 4% PFAand incubated with a blocking/permeabilization buffer [0.3% Triton-X, 10% bovine serum albumin, and 10% goat serum in phosphate-buffered saline (PBS)]. Cells were then incubated with primary antibody overnight at 4°C. Cells were then rinsed three times in PBS and incubated with secondary antibody for 1 h at room temperature. After rinsing three times in PBS and counterstained with 300 ng/mL 4′,6-diamidino-2-phenylindole (DAPI), a single wash was repeated and cells were left in PBS. Cells were viewed under confocal microscopy. The following antibodies were used: Anti-cytokeratin (10 μg/mL; C2931, Sigma), anti-vimentin (10 μg/mL; V6630, Sigma), anti-beta III tubulin (10 μg/mL; MAB1637, Millipore), anti-OCT4 (1:100; sc-8628, Santa Cruz), anti-FGF2R (1:100; sc-122, Santa Cruz), anti-histone 3 phosphoserine 10 (1:500; ab32107, Abcam), anti-mouse immunoglobulin G (IgG; R6393, Invitrogen), and anti-rabbit IgG (1:500; 711-025-152, Jackson Immunoresearch).
Cell cycle synchronization of iPSCs
To synchronize iPSCs, cultures were treated with 200 ng/mL of nocodazole (Sigma) for 16 h. Cells were then released by washing with PBS and replacing with growth medium. To assess the proportion of cells in S phase (DNA synthesis) and M phase prior and after synchronization, the cells were first stained with the Click-iT EdU Imaging Kit (Invitrogen) followed by immunostaining against the M-phase marker H3 phosphoserine 10, respectively. Briefly, iPSCs were given an EdU pulse of 30 min, trypsinized, washed and resuspended in PBS, transferred onto slides by cytospin at 190 rpm for 3 min, fixed in 4% PFA for 15 min at room temperature, and stored in PBS at 4°C. The cells were then stained for EdU according to the manufacturer's instructions, followed by immunofluorescence staining against the M-phase marker as described above.
Reverse-transcription polymerase chain reaction
Total RNA was extracted from blastocysts and cells using the Absolutely RNA Nanoprep Kit (Agilent Technologies) and the RNeasy Mini Kit (Qiagen), respectively. Then, first-strand cDNA was synthesized with SuperScript™ III Reverse Transcriptase (Invitrogen). The QIAGEN® Fast Cycling PCR Kit was used to amplify target cDNA following the manufacturer's instructions. Primers (Table 1) were used at 300 nM. Amplification was carried out in a Mastercycler pro instrument (Eppendorf) with the following program: 45 cycles (35 cycles for GAPDH) at 96°C for 10 sec, 60°C for 30 sec, and 68°C for 30 sec, with an initial activation step of 5 min at 95°C and a final extension step at 72°C for 1 min. The PCR products were resolved in 1.5% agarose gels.
Chromosome spreads
To increase the number of mitotic cells, demecolcine (Wako) was added to a final concentration of 0.1 μg/mL and incubated for 1 h. Cells were trypsinized, centrifuged at 250×g for 4 min, resuspended in 5 mL of ice-cold 0.56% KCl (added dropwise while mixing), and incubated for 10 min at room temperature. Cells were then centrifuged and resuspended with 5 mL of ice-cold fixative (3 parts of methanol to 1 part of glacial acetic acid) added dropwise while vortexing at low speed. Cells were centrifuged, resuspended in 200 μL of fixative, dropped onto slides (prechilled at −20°C), and air-dried. DNA was stained with Fluoroshield/DAPI Mounting Medium. Micrographs were taken with epifluorescence microscopy.
Oocyte maturation, NT, and in vitro fertilization
Ovine ovaries were collected from a local abattoir and transported to the laboratory in warm PBS. Using needle and syringe, medium-sized (2- to 4-mm) follicles were aspirated. Cumulus–oocyte complexes (COCs) with at least two layers of cumulus cells were selected for maturation in in vitro maturation (IVM) medium [bicarbonate-buffered M199 medium (Gibco) supplemented with 10% heat-inactivated FBS, 0.05 U/mL follicle-stimulating hormone/luteinizing hormone (FSH/LH; Pluset), 1.0 μg/mL 17β-estradiol, 50 μg/mL gentamicin, 0.9 mM sodium pyruvate, and 0.1 mM cysteamine). COCs were incubated in a humidified incubator at 39°C and 5% CO2 in air.
NT was performed as described previously (Choi et al., 2010; Lee and Campbell, 2006). Briefly, oocytes were denuded of cumulus cells at 14 h of maturation by treatment with hyaluronidase (0.9 mg/mL, Sigma) and gentle pipetting. Oocytes with extruding spindles at anaphase I/telophase I (AI/TI) were selected for enucleation. These oocytes were incubated in 10 μg/mL Hoechst 33342 and 7.5 μg/mL cytochalasin B in IVM medium for 5 min, and enucleations were carried out in calcium-free HEPES-buffered synthetic oviduct fluid (hSOF) with 7.5 μg/mL cytochalasin B using a micromanipulator. Enucleated oocytes were returned to IVM medium containing 10 mM caffeine for 6 h until cell transfer. Because serum starvation caused extensive cell death and differentiation in iPSCs, cells were first synchronized in M phase with 200 ng/mL nocodazole for 16 h and then in G1 by releasing the cells for 2 h in growth medium without nocodazole. Cells were then collected by trypsinization and resuspended in hSOF.
The cells were kept at 4°C to slow down their metabolism and maintain cell cycle synchrony. PFFs were synchronized in G0 by serum starvation for at least 2 days prior to NT. For cell transfer, one cell was placed into the perivitelline space of each enucleated oocyte and fused with one AC pulse of 1 V for 5 sec and two DC pulses of 100 V for 15 μsec in fusion medium (0.3 M mannitol, 0.1 mM magnesium sulfate, 280 mOsm). Reconstructed embryos were further cultured in IVM medium for 2 h. Embryos were then activated at 25–27 hpm in hSOF containing 5 μM calcium ionophore (A23187) and calcium ions for 5 min, rinsed, and incubated in SOF medium (7.3 mM sodium pyruvate, 0.20 mM
For IVF, ram semen was incubated in 3 mL of capacitation medium (0.33 mM sodium pyruvate, 1.0 mM
Results
Characterization of bFGF/activin-dependent ovine iPSCs
The ovine iPSCs used in this study were generated by forced overexpression of the four Yamanaka factors (Oct4, Sox2, c-Myc, and Klf4) in ovine fetal fibroblasts using retroviral transduction. These iPSCs displayed several characteristics associated with reprogramming toward pluripotency, including formation of compact colonies in culture (Fig. 1A), downregulation of the fibroblast marker FSP1, and upregulation of endogenous SOX2 and OCT4 (Fig. 1B), and positive AP staining (Fig. 1G). OCT4 was also detected by immunostaining together with FGFR2, a receptor for bFGF (Fig. 1C). Surprisingly, iPSCs were negative for NANOG (Fig. 1B). Expression of the TE markers GATA3 and CDX2 were not observed in either iPSCs or PFFs, whereas they were detected in IVF blastocysts (Fig. 1B). COL1A1, another gene highly expressed in fibroblasts, was also expressed in iPSCs (Fig. 1B), but expression levels were 70 times higher in the PFFs as estimated by quantitative RT-PCR (not shown). Ovine iPSCs also expressed the exogenous reprogramming factors (Fig. 1D). At passage 40, metaphase spread analysis of iPSCs revealed that of the 50 spreads counted, all were diploid and 68% displayed a normal karyotype with 54 chromosomes (Fig. 1E), with the remaining 32% showing a range of aneuploidies (between 50–55 chromosomes). The parental cells had a normal karyotype.
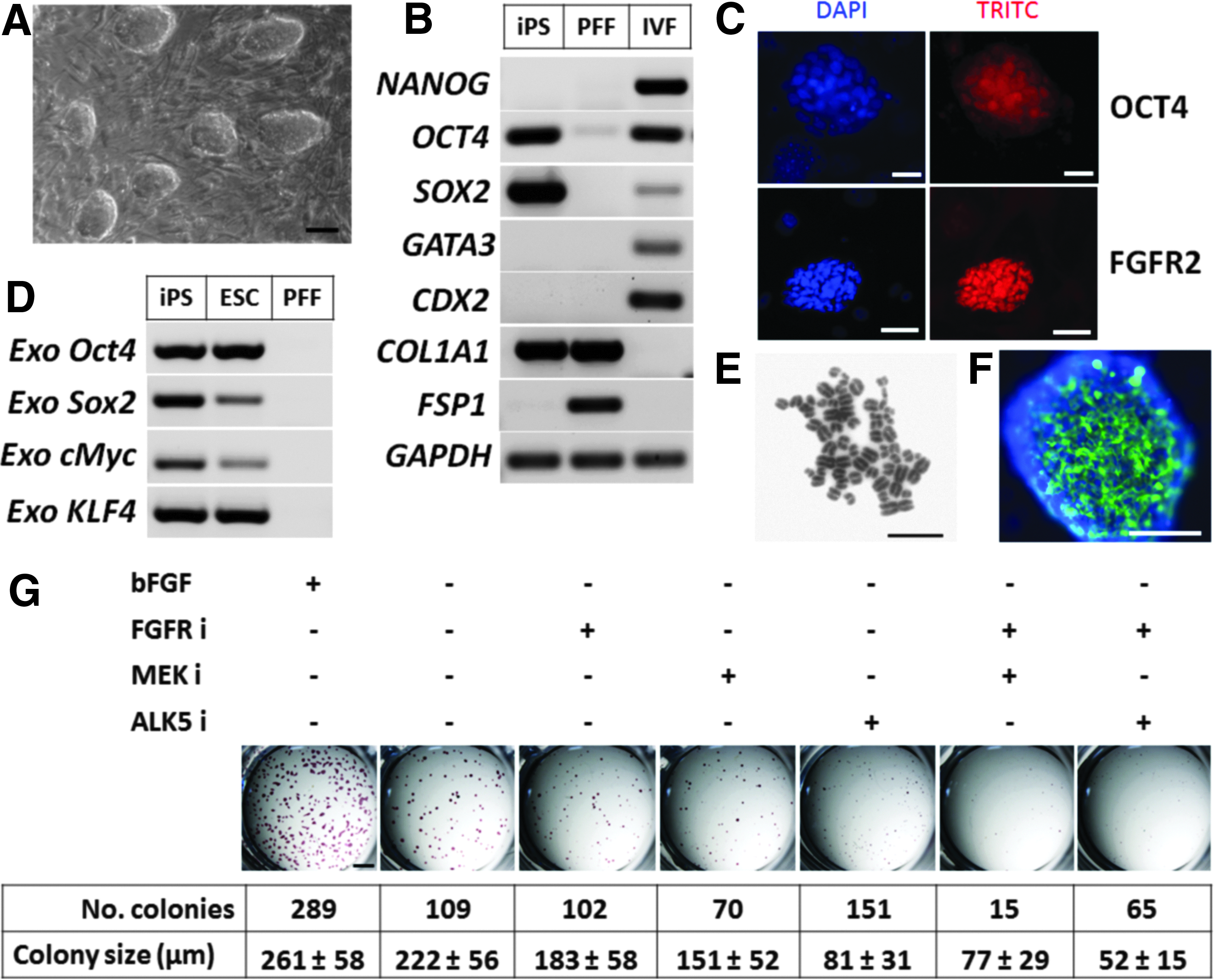
Characterization of ovine iPSCs.
Because bFGF was supplemented to the medium during the generation of these iPSCs and subsequent culture, we wished to determine whether the iPSCs required bFGF for self-renewal and proliferation. Furthermore, the cells were grown on a feeder layer of MEFs, a source of activin A that helps iPSCs maintain pluripotency by signaling through activin receptor-like kinase (ALK) receptors (Beattie et al., 2005). To study the FGF and activin A requirements of these iPSCs, we carried out a colony-formation assay by seeding the cells at low density and determined their AP activity after 10 days culture (Fig. 1G). Removing bFGF from the medium reduced the number of colonies (38%, total) as well as their average size (85%) relative to control (100%). Colony formation in bFGF-deprived medium was further decreased by inhibiting MEK signaling (24%) and still further with double inhibition of MEK and FGFRs (5%) relative to control. The size of bFGF-deprived iPSC colonies was progressively reduced by FGFR inhibition (70%), MEK inhibition (58%), ALK5 receptor inhibition (31%), FGF2R and MEK double inhibition (30%), and FGFRs and ALK5 double inhibition (20%) relative to control. We further confirmed the activin A requirements of these iPSCs because they stained positive for the ectodermal marker βIII tubulin after inhibition of ALK5 receptors for 10 days (Fig. 1F). Taken together, this assay showed that these ovine iPSCs required bFGF and that self-renewal and proliferation were regulated by FGF/ERK together with activin A signaling pathways.
To test whether these ovine iPSCs were pluripotent, we assessed their capacity to differentiate in vitro into cell lineages derived from the three somatic germ layers. Under conditions inducing differentiation (see Materials and Methods), the cells aggregated into EBs, and, after plating, they differentiated into cells positive for α-fetoprotein (endoderm), vimentin (mesoderm), and βIII tubulin (ectoderm) (Fig. 2).
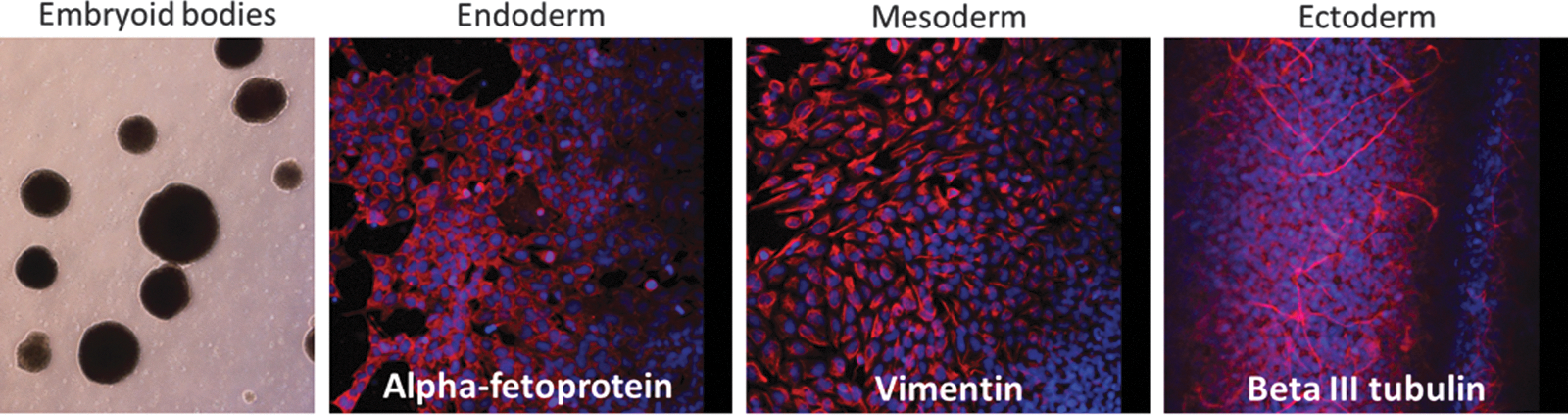
Differentiation capacity of iPSCs in vitro. iPSCs were induced to form EBs under permissive conditions and then plated. Then, the differentiated cells were immunostained for markers representing the three somatic germ layers. Color images available online at www.liebertpub.com/cell
Cell cycle synchronization of ovine iPSCs
We used EDU labeling and anti-phosphohistone H3 immunostaining to determine the cell cycle profile of iPSCs. In continuous iPSCs cultures, 40% of cells were in S phase whereas 5% were in mitosis, with the remaining 55% in G1 and G2. We first attempted to carry out NT with cells that had been synchronized in M phase by a short block with demecolcine followed by “shake off” to release the mitotic cells (Kasinathan et al., 2001). This preliminary data showed that development to blastocysts with M-phase donors was very low, 4% for PFF-NT and 0% for iPSC-NT. Therefore, we opted to synchronize the iPSCs in G1 using nocodazole, because synchronization by serum starvation to G1/G0 resulted in extensive cell death and differentiation. To this end, we synchronized the cells in M phase by nocodazole block for 16 h and then released them (Fig. 3A, B). After 16 h of blocking with 200 ng/mL nocodazole, the proportion of iPSCs in mitosis was enriched to 55%, while the proportion of cells in S-phase was halved (20%). After 2 h release from nocodazole, these mitotic cells had progressed into G1 because only 4% of cells remained in M and 9% were in S phase. iPSCs entered S phase between 4 h and 6 h postrelease, with about 66% of cells progressing from G1 to S phase between 4 h and 10 h postrelease (Fig. 3A, B). These data show that the proportion of ovine iPSCs in G1 can be enriched by nocodazole block and release. After 2 h release, we selected small iPSCs as presumptive G1 donors for NT (Fig. 3C).

Synchronization of ovine iPSCs in G1.
Ovine iPSCs produce fewer blastocysts than PFFs after NT
After synchronization of iPSCs in G1 and of PFFs in G0 prior to NT, we determined the preimplantation development of the reconstructed embryos (Table 2). Results from three replicates show that cleavage rate was high in both groups (79% for iPSC-NT and 90% for PFF-NT); however, blastocyst formation was significantly lower with iPSC-NT (2%) compared to PFF-NT (10%, p<0.01, chi-squared analysis).
Determined after 3 and 8 days from activation, respectively. p<0.01 chi-squared analysis.
iPSC, induced pluripotent stem cell; PFF, parental fetal fibroblast.
iPSCs are not fully reprogrammed following NT
To assess nuclear reprogramming in reconstructed embryos with different donor cells, we analyzed the expression of NANOG, GATA3, and CDX2 at the blastocyst stage by end-point RT-PCR. We also included SOX2 in the analysis to determine whether this gene, which is highly expressed in iPSCs, was downregulated in iPSC-NT blastocysts. Analysis was carried out in three biological replicates for both groups together with IVF controls (Fig. 4A). We did not detect SOX2 in any of the IVF blastocysts, whereas expression was observed in 2/3 of replicates in iPSC-NT blastocysts and 1/3 in PFF-NT blastocysts (Fig. 4B, C). The TE markers GATA3 and CDX2 were consistently activated in all replicates (Fig. 4B). Surprisingly, NANOG was not activated in 2/3 iPS-NT blastocysts, whereas IVF and PFF-NT blastocysts activated this pluripotency gene in all three replicates (Fig. 4B, C).
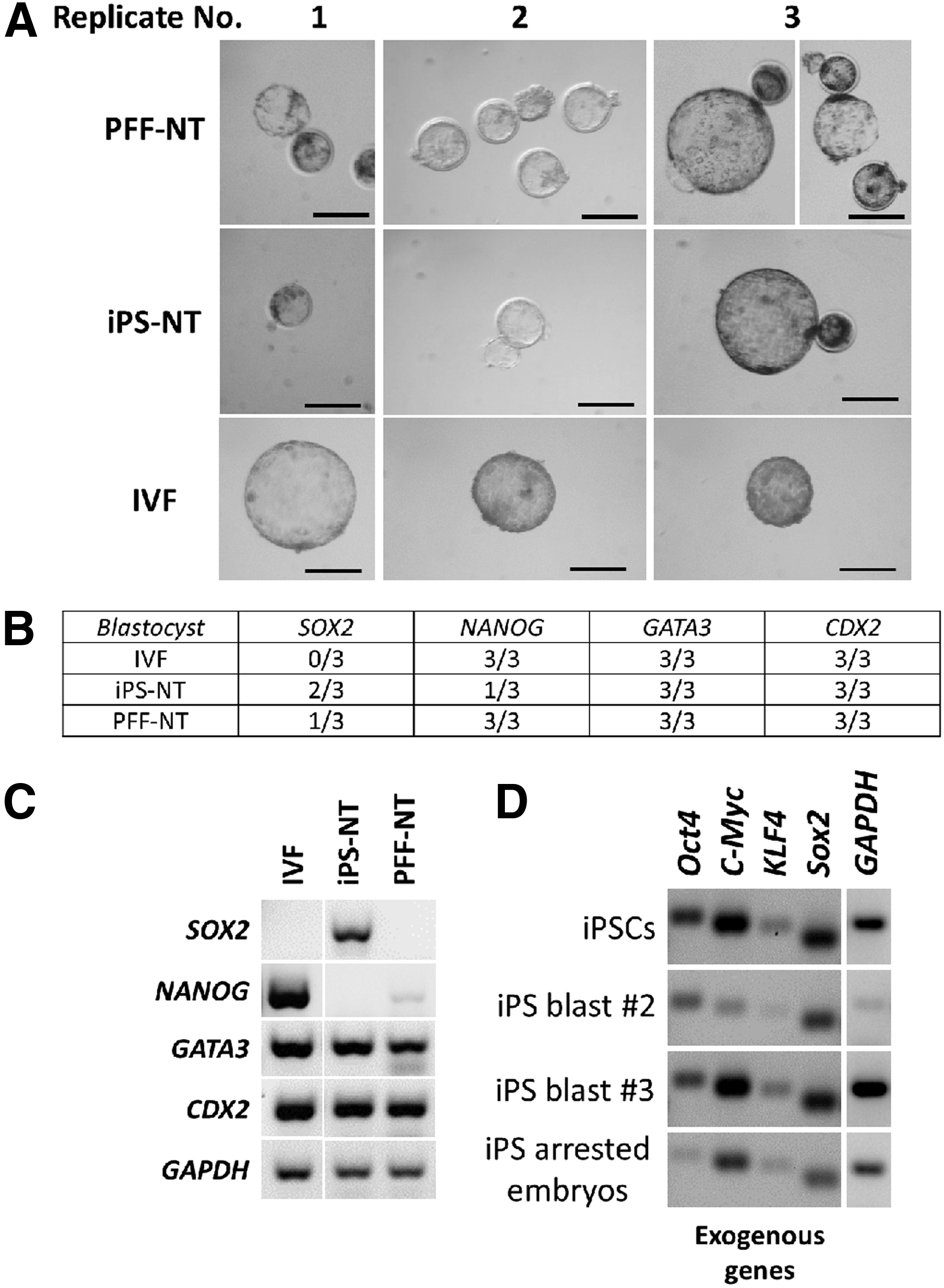
Detection of pluripotent and TE markers as well as exogenous genes by PCR.
We also investigated whether the exogenous pluripotency genes expressed in iPSCs were silenced following NT. We found that all four exogenous genes were still expressed both at the blastocyst stage and in embryos that failed to form blastocysts (Fig. 4D).
Discussion
In this study, we investigated the growth characteristics and developmental capacity of bFGF-dependent ovine iPSCs. We confirmed the dependency of these cells on bFGF and activin A signaling for self-renewal and proliferation, similar to the signaling requirement of hESCs and epiblast stem cells (Alberio et al., 2010; Brons et al., 2007; Tesar et al., 2007; Vallier et al., 2005). However, in contrast to hESCs and epiblast stem cells, which form flat colonies and usually do not tolerate passaging by trypsin, our ovine iPSCs formed dome-shaped colonies and were passaged as single-cell suspensions. In this respect, ovine iPSCs were more similar to mouse ESCs. These characteristics are consistent with ovine iPSCs reported in previous studies (Bao et al., 2011; Liu et al., 2012; Sartori et al., 2012).
Nonetheless, using a colony-formation assay, we showed that our ovine iPSCs were bFGF and activin A dependent. Removing bFGF from the culture medium resulted in fewer and smaller colonies, and this was further confirmed using FGF/ERK signaling inhibitors. Furthermore, inactivation of the ALK5 receptors with a specific inhibitor had a more pronounced effect at reducing proliferation. This is consistent with previous reports showing that the activin A/Nodal pathway is required to maintain self-renewal of mouse epiblast stem cells (Brons et al., 2007; Tesar et al., 2007) as well as pluripotency in pig epiblast stem cells (Alberio et al., 2010; Park et al., 2013). Double inhibition of FGFRs and MEK or FGFRs and ALK5 had a synergistic effect on proliferation compared to either inhibitor alone. Taken together, these functional data support that both FGF/ERK and activin A transduction signaling pathways are important for self-renewal of ovine iPSCs.
Persistent expression of transgenes has been reported in iPSC lines from pigs (Esteban et al., 2009; Ezashi et al., 2009; Fan et al., 2013; Rodriguez et al., 2012; West et al., 2010), cattle (Sumer et al., 2011), mice (Okita et al., 2007), humans (Liu et al., 2009; Ramos-Mejia et al., 2012), monkeys (Liu et al., 2008), and sheep (Sartori et al., 2012). Moreover, it appears that overexpression of the transgenes is necessary to maintain pluripotency in iPSCs of farm animals because differentiation has been reported following inactivation of the exogenous factors in doxycycline (DOX)-inducible systems (Bao et al., 2011; Hall et al., 2012; Li et al., 2011; Rodriguez et al., 2012).
This continued dependency on expression of exogenous reprogramming factors suggests incomplete activation of the network of endogenous genes associated with pluripotency. Although both ovine studies involving DOX-inducible systems reported expression of endogenous OCT4, SOX2, and NANOG (Bao et al., 2011; Li et al., 2011), it is possible that other genes associated with pluripotency were not activated or that expression levels of endogenous pluripotent genes were low and thus insufficient to maintain pluripotency without expression of the transgenes. We found that endogenous OCT4 and SOX2 were expressed whereas NANOG was not, suggesting incomplete reprogramming of the pluripotency gene network. The lack of NANOG expression was consistent in multiple iPSC lines generated (not shown). Although NANOG is essential for pluripotency in the embryo (Chambers et al., 2003), recent evidence shows that iPSCs with full differentiation capacity can be generated without Nanog (Carter et al., 2014; Schwarz et al., 2014), suggesting that the four-factor overexpression system can bypass the requirement of Nanog during iPSC reprogramming. In agreement with these reports, ovine iPSCs were capable of differentiating into cells derived from the three germ layers in vitro, demonstrating that they were pluripotent. However, we found that NANOG reactivation was very limited in iPSC cloned blastocysts, suggesting that these iPSCs were locked in a reprogrammed state resistant to the reprogramming machinery of the oocyte.
Despite the synchronization of iPSCs in G1, we found that development to blastocysts of cloned embryos was not improved compared to those produced using fibroblasts. This finding is consistent with a recent publication reporting similar developmental competence of pig iPSCs and fibroblasts (Xie et al., 2014). While this manuscript was in preparation, another report showed that iPSCs synchronized in G2/M are better suited for NT compared to G1 and unsynchronized cells (Yuan et al., 2014). Improved development with G2/M-synchronized donors was also reported with mouse iPSCs (Kou et al., 2010). However, the general observation with G2/M-phase donors is that the resulting embryos have an increased proportion of aneuploidies, and therefore, are unlikely to support full-term development.
We next sought to determine features of genetic reprogramming in cloned embryos produced with different cell types. We found that blastocysts produced with PFFs are better reprogrammed than those produced with iPSCs. NANOG, which was silent in both iPSCs and PFFs, remained inactive in most iPSC-NT blastocysts, whereas it was consistently expressed in PFF-NT and IVF blastocysts. This was surprising because it was expected that a gene associated with pluripotency would be activated more easily in iPSCs than in differentiated cells. SOX2, in contrast, was not expressed in any of the IVF blastocysts, whereas it was expressed in 2/3 iPSC-NT blastocysts and in 1/3 of PFF-NT blastocysts. This suggests that SOX2 was not properly silenced following iPSC-NT.
It was surprising not to detect activation of SOX2 in IVF blastocysts because we had previously detected low levels in control IVF blastocysts when characterizing gene expression of the cells (Fig. 1). One possibility for this incongruence is that SOX2 starts to be expressed at this stage at low levels in some blastocysts, so it could be detected in a pool of ten blastocysts (Fig. 1) but not in individual blastocysts (Fig. 4). Alternatively, culture conditions might have affected gene expression because the IVF control embryos used in Figure 1 had been cultured with BSA whereas the ones in Figure 4 were cultured with FBS (as were the cloned embryos). As with endogenous SOX2, we observed no silencing of exogenous pluripotency genes during preimplantation development of sheep iPSC-NT embryos, which is consistent with the recent publication regarding pigs (Xie et al., 2014). Activation of TE lineage markers was normal because both iPSC-NT and PPF-NT blastocysts expressed CDX2 and GATA3. This is consistent with a previous study in mice, which showed CDX2 expression in the TE of iPSC-NT blastocysts by immunocytochemistry (Kou et al., 2010). With the exception of the TE lineage, our data support that genetic reprogramming in iPSCs is incomplete following NT. The incomplete reprogramming of iPSCs after NT attests to the poor quality of iPSC-NT embryos and could explain the low development of cloned iPSC embryos reported here and in a previous study (Fan et al., 2013). To our knowledge, this is the first study comparing gene expression reprogramming of key developmental genes between cloned blastocysts produced with iPSCs versus parental fibroblast cells.
In summary, our data support that reprogramming of ovine iPSCs after NT is incomplete. This might be an important factor explaining the low developmental capacity of reconstructed embryos with iPSCs in farm animals. Persistent expression of transgenes and increased number of aneuploidies in donor cells may be the reason for compromised reprogramming and developmental potential NT embryos, although the underlying reason for these has yet to be determined. Nonetheless, from our results in sheep and from those reported by other groups in pigs (Fan et al., 2013; Xie et al., 2014), it is plausible to propose that iPSCs generated by nonintegrating methods (free of transgenes) would be better candidates for the production of transgenic animals by NT.
Footnotes
Author Disclosure Statement
The authors declare that no conflicting financial interests exist.