
Abstract
Abstract
Xeroderma pigmentosum group A (XP-A) is a genetic disorder in which there is an abnormality in nucleotide excision repair that causes hypersensitivity to sunlight and multiple skin cancers. The development of central and peripheral neurological disorders not correlated to ultraviolet light exposure is associated with XP-A. The genes responsible for XP-A have been identified and a XPA knockout mouse has been generated. These knockout mice exhibit cutaneous symptoms, but they do not show neurological disorders. The mechanism of pathogenesis of neurological disorders is still unclear and therapeutic methods have not been established. Therefore, we generated XP-A patient-derived human induced pluripotent stem cells (XPA-iPSCs) to produce in vitro models of neurological disorders. We obtained iPSC lines from fibroblasts of two patients carrying different mutations. Drugs screened using XPA-iPSC lines can be helpful for treating XP-A patients in Japan. Additionally, we revealed that these iPSCs have the potential to differentiate into neural lineage cells, including dopaminergic neurons, which decrease in XP-A patients. Our results indicate that expression of the normal XPA gene without mutations is not required for generation of iPSCs and differentiation of iPSCs into neural lineage cells. XPA-iPSCs may become useful models that clarify our understanding of neurological pathogenesis and help to establish therapeutic methods.
Introduction
X
In Japan, approximately 0.001% of the population is affected by XP (Moriwaki et al., 2012), with the XP-A group as the most common form (Moriwaki et al., 2012). The cells derived from XP-A patients are unable to repair DNA damage induced by UV light. Therefore, they are photosensitive and have a higher predisposition to various skin cancers. The incidence of skin cancer in XP patients is about 1000-fold greater than that for healthy individuals; from a young age, XP-A patients commonly develop cutaneous symptoms, such as pigmented spots (Moriwaki et al., 2012). In addition, XP-A patients show central and peripheral neurological disorders, such as sensory hearing loss, mental retardation, cerebellar ataxia, extrapyramidal abnormalities, rigidity, dysphagia, and laryngeal dystonia along with cutaneous symptoms (Hayashi et al., 2004; Miyata et al., 2010).
These neurological disorders develop independently of UV light exposure, and their pathogenic mechanisms are unknown (Brooks, 2007). Currently, there are no therapies for treating the various neurological disorders of XP-A patients. Knockout mice for the XP-A group have been established (de Vries et al., 1995; Nakane et al., 1995); these mice are extremely sensitive to UV light, develop skin cancers easily, and are useful models of cutaneous symptoms (de Vries et al., 1995; Nakane et al., 1995). However, XPA knockout mice do not develop neurological disorders (Nakane et al., 1995).
To clarify the pathogenic mechanisms and establish therapies for XP-A neurological disorders, a new disease model is required. Human-induced pluripotent stem cells (hiPSCs) were established by two groups in 2007 (Takahashi et al., 2007), making it possible to create new disease models by generating iPSCs from patient-derived cells (Egashira et al., 2013). In the current study, we sought to generate in vitro models of XP-A neurological disorders and investigate their potential to differentiate into neural lineage cells.
Materials and methods
This research study was approved by the ethics committee of the National Institute of Advanced Industrial Science and Technology.
Cell cultures
We purchased human dermal fibroblasts (HDFs) from Cell Applications (San Diego, CA, USA). The XP-A patient-derived fibroblast lines XP3OS (Satokata et al., 1990) and XP39OS (Sato et al., 1987; Satokata et al., 1992) were donated by the Japanese Collection of Research Bioresources Cell Bank (JCRB). HDFs were maintained in Dulbecco's modified Eagle medium (DMEM; Invitrogen, Carlsbad, CA, USA) containing 10% fetal bovine serum (FBS; JRH Biosciences, KS), 100 U/mL penicillin, and 100 μg /mL streptomycin (Invitrogen). XP3OS cells were maintained in Eagle's minimum essential medium (EMEM; Invitrogen) containing 10% FBS (JRH), 100 U/mL penicillin, and 100 μg/mL streptomycin (Invitrogen). XP39OS cells were maintained in α-MEM (Invitrogen) containing 20% FBS (JRH), 100 U/mL penicillin, and 100 μg/mL streptomycin (Invitrogen). The iPSCs we generated were maintained in human embryonic stem cell (hESC) medium (DMEM/F-12 with GlutaMAX-I; Invitrogen) containing Knockout Serum Replacement (KSR; Invitrogen), 0.1 mM nonessential amino acids (Invitrogen), 0.1 mM 2-mercaptoethanol (Invitrogen), 100 U/mL penicillin, 100 μg/mL streptomycin, and 5 ng/mL recombinant human basic fibroblast growth factor (bFGF; WAKO, Osaka, Japan). We passaged iPSCs using dissociation solution (0.25% trypsin, 0.1 mg/mL collagenase type IV, 10 mM CaCl2, and 20% KSR in distilled water).
Generation of iPSCs
Plasmid generation and retrovirus production were carried out as previously described (Aoki et al., 2010). The XP3OS, XP39OS, and HDF cells were seeded onto 100-mm culture dishes (5×104 cells/100 mm) and cultured overnight. The culture medium was then exchanged with retrovirus-containing supernatant and cells were cultured for 24 h; this step was repeated once. The culture supernatant was removed, and DMEM was supplemented with 15% FBS. The culture medium was then changed daily. After 3 days, retrovirus-infected cells were seeded onto SNL feeder cells (5×103 to 5×105 cells/100-mm dish). The next day the medium was changed to hESC medium and refreshed every other day for 4 weeks. Around 25 days postinfection (dpi), colonies were selected on the basis of whether their morphology was similar to that for hESC-like colonies. Selected colonies were subsequently expanded and maintained on SNL feeder cells in hESC medium.
RNA isolation and reverse transcription polymerase chain reaction
Total RNA was isolated using an RNeasy Mini Kit (QIAGEN, Hilden, Germany). We used 1 μg of RNA to synthesize complementary DNA (cDNA), in conjunction with a ReverTra Ace-α™ Kit (Toyobo, Osaka, Japan) and oligo(dT)20 primers. We conducted PCRs using an ExTaq HS™ Kit (Takara Bio, Shiga, Japan) and specific primers (Table 1).
Alkaline phosphatase staining
Alkaline phosphatase (ALP) staining was performed using a Leukocyte ALP Kit (Sigma-Aldrich, Tokyo, Japan) according to the manufacturer's instructions.
Immunofluorescence
Cells were fixed with phosphate-buffered saline (PBS) containing 4% paraformaldehyde for 10 min and permeabilized with 0.1% Triton X-100 for 10 min at room temperature. Cells were then treated with 1% (wt/vol) bovine serum albumin (BSA; Sigma) for 10 min at room temperature. Cells were incubated with the appropriate primary antibody overnight at 4°C. Cells were then incubated with the appropriate secondary antibody for 30 min at room temperature. The primary antibodies we used were against: stage-specific embryonic antigen-3 (SSEA-3; MAB4303, 1:200; Millipore, Billerica, MA, USA), SSEA-4 (MAB4304, 1:200, Millipore), tumor-related antigen-1-60 (TRA-1-60; ab16288-200, 1:200, Abcam, Cambridge, UK), TRA-1-81 (ab16289-200, 1:200, Abcam), OCT4 (ab19857-100, 1:200, Abcam), NANOG (ab21624,1:50,,Abcam), SOX17 (AF1924,1:200,R&D Systems, Minneapolis, MN, USA), α-smooth muscle actin (α-SMA; N1584, prediluted, Dako, Glostrup, Denmark), βIII-tubulin (CBL412, 1:200, Millipore), and tyrosine hydroxylase (AB152, 1:500, Millipore). The secondary antibodies we used were AlexaFluor-conjugated antibodies from Invitrogen, and they were diluted 1:300. Nuclei were visualized using 0.2 μg/mL Hoechst 33342 (Molecular Probes, Eugene, OR, USA).
In vitro differentiation
For embryoid bodies (EBs) formation, hESC-like colonies were cultured in low-attachment culture dishes (Prime Surface; Sumitomo Bakelite, Tokyo, Japan) in hESC medium for 12 days, with culture medium changed every other day. EBs were then seeded onto gelatin-coated plates for an additional 12 days of culture in the same medium.
Differentiation into neuronal cells
EBs derived from wild-type iPSCs and XPA-iPSCs were seeded onto plates coated with ornithine and fibronectin. Attached EBs were cultured, differentiated, and characterized using a Neural Identification Kit (R&D) according to the manufacturer's instructions.
Differentiation of iPSCs into dopaminergic neurons was performed using the stromal cell–derived inducing activity (SDIA) method, as previously described (Kawasaki et al., 2000). Undifferentiated iPSC colonies were seeded onto PA6 feeder cells cultured in Glasgow's minimum essential medium (GMEM; Invitrogen) containing 10% KSR, 0.1 mM MEM-nonessential amino acids, 0.1 mM 2-mercaptoethanol, 100 U/mL penicillin, and 100 μg/mL penicillin, for 12 days.
Results
Generation of XPA-iPSCs
Around 25 dpi with retrovirus, we obtained several hESC-like colonies. We selected one ESC-like colony from each cell line and allowed these to expand over several passages (Fig. 1A). All colonies showed high ALP activity (Fig. 1A). The human ESC-like clones that we generated from XP3OS, XP39OS, and HDF were designated XP3R, XP39R, and HDFR.
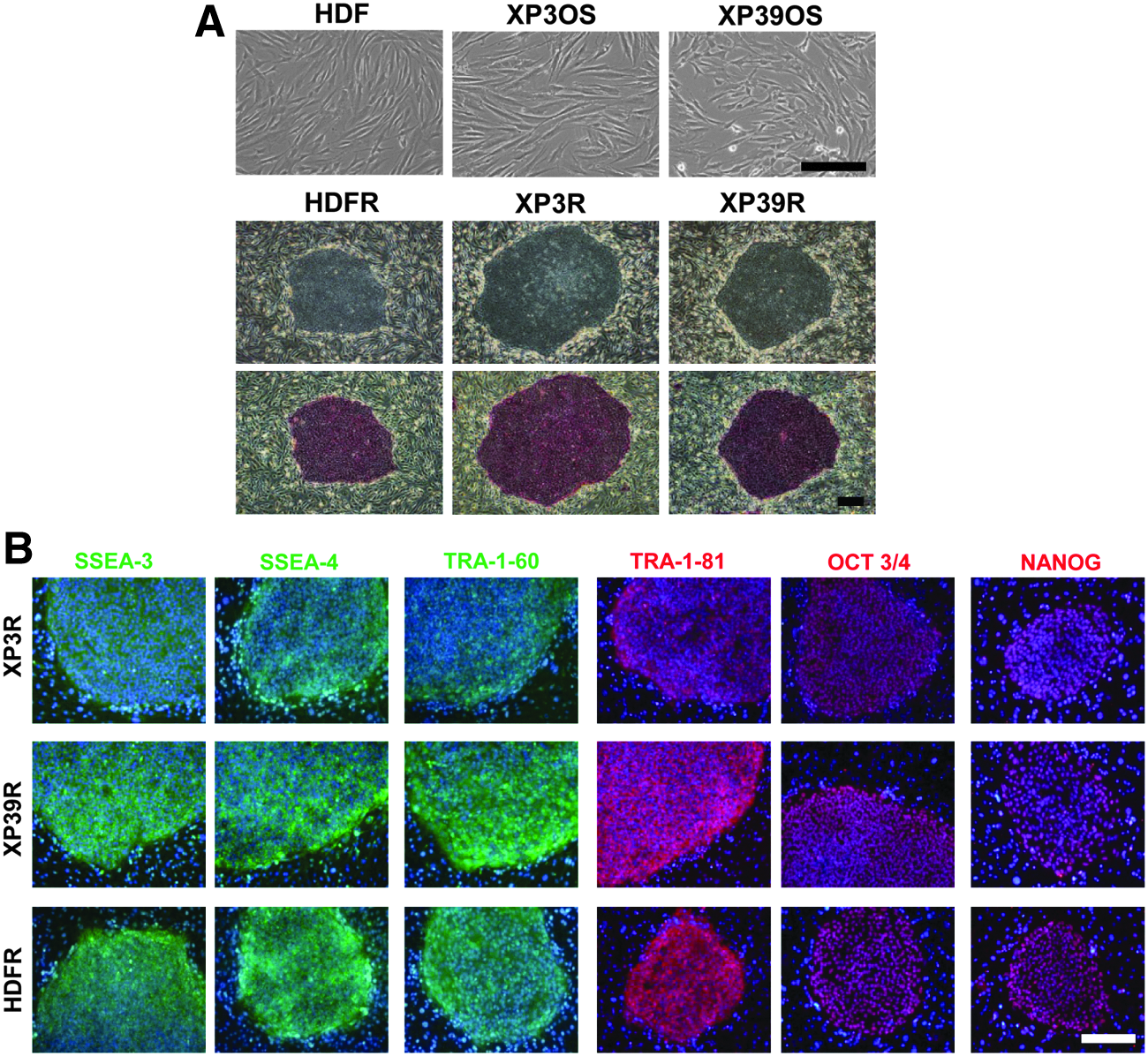
Morphology of XP3OS, XP39OS, and HDF parental cells and ESC-like colonies (XP3R, XP39R, and HDFR, respectively). (
Characterization of XPA-iPSCs
To verify that colonies derived from XP-A patient fibroblasts were authentic iPSCs, we examined the expression of pluripotent markers using immunofluorescence and reverse transcription polymerase chain reaction (RT-PCR) assays.
Immunofluorescence assays showed that all colonies expressed the cell-surface antigens SSEA-3, SSEA-4, TRA-1-60, and TRA-1-81, along with the transcription factors OCT3/4 and NANOG (Fig. 1B). Our RT-PCR results revealed that colonies expressed the pluripotent maker genes OCT3/4, SOX2, NANOG, REX1, UTF1, GDF3, DPPA2, DPPA4, and DPPA5. The retroviral transgenes (Tg) should be silenced in reprogrammed cells. In those colonies, Tg-OCT3/4, Tg-SOX2, Tg-KLF4, and Tg-c-MYC were not expressed. We detected expression of KLF4 and c-MYC in all samples (Fig. 2).
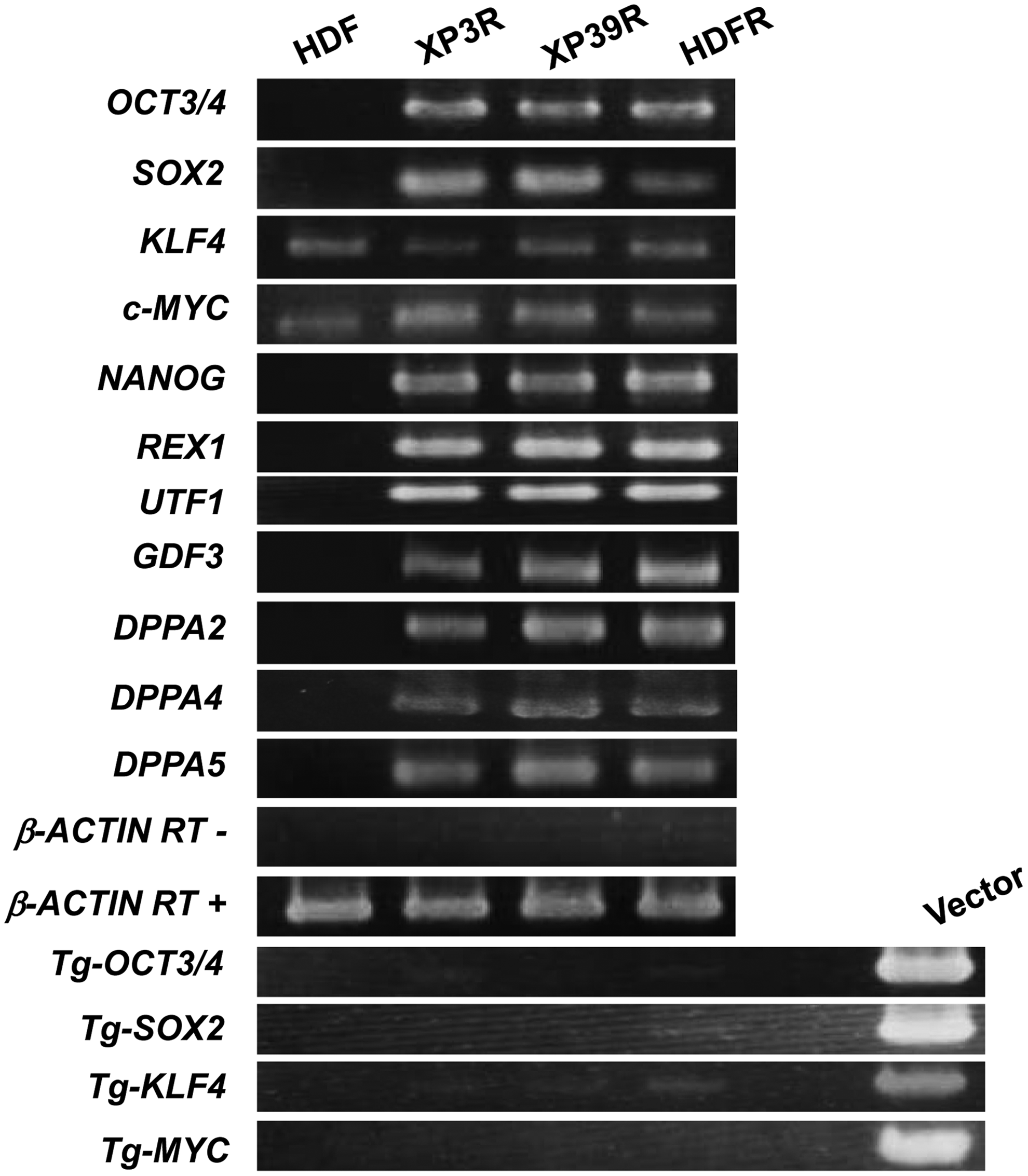
Examination of expression of pluripotent marker genes, endogenous KLF4 and c-MYC, and retroviral transgenes (Tg).
Confirmation of mutations in XPA-iPSCs
We confirmed the presence of mutations in our XPA-iPSCs through sequencing analyses (Fig. S1 and Supplementary Text) (Supplementary Data are available at www.liebertpub.com/cell/). Each XPA-iPSC line had parental cell–derived mutations, as previously reported (Maeda et al., 1994; Satokata et al., 1990). XP3R had a mutation at the 3′ splice acceptor site of intron 3 (TTTCA
Differentiation potential of XPA-iPSCs
After 12 days of floating culture, dissociated cells from ESC-like colonies formed EBs. These EBs were seeded onto gelatin-coated plates and cultured for an additional 12 days. Immunofluorescence showed that these cells were positive for the expression of βIII-tubulin (ectodermal marker), α-SMA (mesodermal marker), and SOX17 (endodermal marker; Fig. 3A). Analysis by RT-PCR confirmed that cells expressed MAP2, PAX6 (endodermal marker), TNTC (mesodermal marker), SOX17, and AFP (endodermal marker; Fig. 3B). The ESC-like colonies had the potential to differentiate into cells of the three germ layers in vitro and were authentic iPSCs with pluripotency.

In vitro differentiation of ESC-like colonies. (
We also seeded EBs onto plates coated with ornithine and fibronection. After 4 days, cells were positive for NESTIN (Fig. 4a). After 14 days in differentiation medium, cells were positive for the neuronal marker βIII-tubulin (Fig. 4b) or the glial marker glial fibrillary acidic protein (GFAP) (Fig. 4c). For dopaminergic differentiation, XPA-iPSCs were seeded onto PA6 stromal cells and cultured under differentiation conditions for 2 weeks (Kawasaki et al., 2000). Some of the XPA-iPSCs had neuronal structures, with some cells positive for βIII-tubulin and tyrosine hydroxylase (Fig. 5A). Expression of marker genes specific for dopaminergic neurons, such as aromatic amino acid decarboxylase (AADC), dopamine transporter (DAT), choline acetyltransferase (ChaT), and microtubule-associated protein 2 (MAP2), was detected using RT-PCR assays (Fig. 5B).

Differentiation of XPA-iPSCs into a neuronal lineage. Immunofluorescent staining of differentiated cells. Nestin (green) (
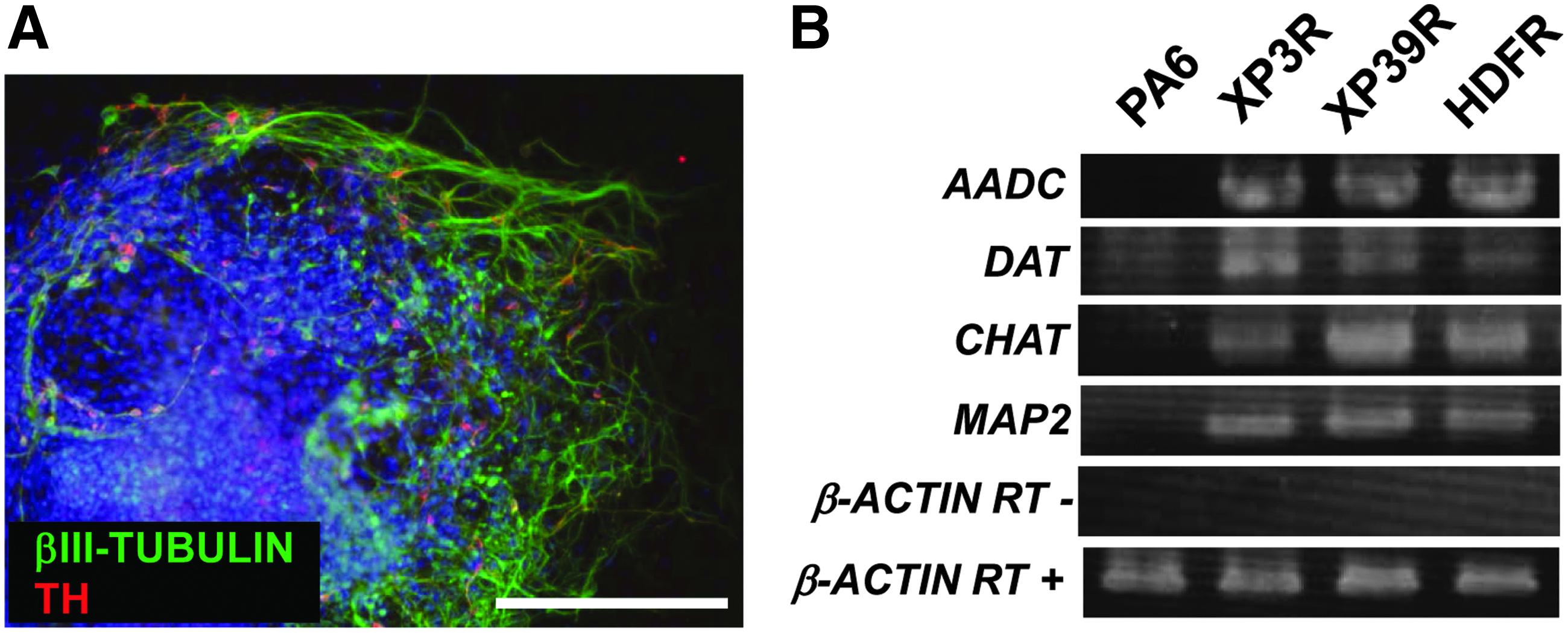
Differentiation of XPA-iPSCs into dopaminergic neurons. (
Discussion
The ability to generate disease-specific iPSCs has made it possible to show the pathogenesis for several diseases, with multiple disease models already established (Egashira et al., 2013) for Rett syndrome and Down syndrome (Wang and Doering, 2012). We generated iPSCs from XP-A patients to establish an in vitro model of XP-A. Recently, direct conversion methods, which are new techniques for generating specific cell lineages from somatic cells without iPSCs, have been developed (Sancho-Martinez et al., 2012), and direct conversion into neuronal lineages has been reported (Son et al., 2011; Yoo et al., 2011). However, because XP-A patients demonstrate neurological disorders and cutaneous symptoms, any generated iPSCs must be able to differentiate into several cell lineages to allow for examination of symptoms in several tissues. It has also been reported that genes related to gene repair play a more significant role in ESCs than in somatic cells (de Waard et al., 2008; Maynard et al., 2008).
We were successful in generating iPSC lines from the fibroblasts of two XP-A patients. Our results suggest that the normal XPA gene without mutation is not required for the generation of iPSCs. One fibroblast line carried the XP3OS mutation; this is a homozygote with a mutation at the 3′ splice acceptor site of intron 3 of the XPA gene. These cells were derived from a patient showing a severe phenotype of cutaneous symptoms and neurological disorders (Maeda et al., 1994). The second fibroblast line carried the XP39OS mutation; this is a homozygote with a nonsense mutation in exon 6 of the XPA gene. These cells were derived from a patient showing a milder phenotype of cutaneous symptoms and neurological abnormalities (Sato et al., 1987; Satokata et al., 1990). The XP3OS genotype is prevalent in around 80% of XP-A patients in Japan (Moriwaki et al., 2012). Therefore, to develop effective therapies, it would appear essential to conduct drug screening using XPA-iPSCs. Additionally, a comparison between the phenotypes of two XPA-iPSC lines is possible because the iPSC line generated from XP39OS is available.
We examined whether XPA-iPSCs could differentiate into cells of a neural lineage and found that they could differentiate into neurons and glia. We showed that they could differentiate into dopaminergic neurons using a specific differentiation method. XP-A patients have neurological disorders affecting the central and peripheral nervous systems. A decrease in the number of dopaminergic neurons in the basal ganglion and brainstems of XP-A patients has been previously reported (Hayashi et al., 2004). Therefore, XPA-iPSC–derived dopaminergic neurons might be good targets for evaluating neurological disorders. The neural tissues of XP-A patients appear to develop normally, although it remains unclear whether these tissues develop fully. It is possible that our neural induction results from XPA-iPSCs mimic the state of neural tissues in XP-A patients.
In future studies, we hope to examine functionally mature XPA-iPSC–derived neurons, along with degenerative processes following synapse formation. We believe that sensory neurons are suitable for this purpose given that sensorineural hearing loss is a sign of the onset of XP-A neurological disorders. Long-term examination of the auditory brainstem response (ABR) in 20 XP-A cases revealed that auditory abnormalities were not observed until patients were 4 years old; a lack of any ABR was seen in patients older than 10 years (Sugimoto et al., 1999). Histological analysis showed auditory nerve degeneration (Viana et al., 2013). These findings show that sensory neurons can develop and form neural networks with hair cells of the inner ear. These neural networks gradually break down over a long period in XP-A patients. Differentiation methods for sensory neurons from hESCs have been reported (Needham et al., 2014; Shi et al., 2007). These hESC-derived neurons were able to form synapses with hair cells in cultured sensory epithelia from the inner ears of mice or rats in vitro (Nayagam et al., 2013). These experiments highlight the potential of XPA-iPSC–derived sensory neurons as models of XPA-related neurological disorders.
Moreover, to culture XPA-iPSC–derived neurons in an environment similar to that experienced in vivo may be required for reproduction of phenotypes of neurological disorders. Cyclopurines formed by oxygen radicals are specifically repaired by NER (Brooks, 2007; Kuraoka et al., 2000). This oxidative damage inhibits DNA replication and the transcriptional response; death of XP-A patient neuronal cells could be due to weak NER activity or its absence (Brooks et al., 2000). Therefore, we plan on using XPA-iPSC–derived neurons cultured under conditions giving rise to free radicals and examining factors related to oxidative stress.
In conclusion, we generated two iPSC lines from two XP-A patients and successfully differentiated both XPA-iPSC lines into neural lineage cells. Our findings indicate that the normal XPA gene without mutation is not required for iPSC generation and neural differentiation. We are confident that the iPSCs generated from XP-A patient fibroblasts in our study can be used as models of neurological disorders in XP-A patients.
Footnotes
Acknowledgments
We thank Dr. Toshio Kitamura for providing the retrovirus system. We also thank our colleagues at the Tissue Engineering Research Group, Health Research Institute, National Institute of Advanced Industrial Science and Technology (AIST). This work was partially supported by the Project for Realization of Regenerative Medicine from the Ministry of Education, Culture, Sports, Science and Technology–Japan (MEXT).
Author Disclosure Statement
The authors declare that there are no conflicts of interest.
References
Supplementary Material
Please find the following supplemental material available below.
For Open Access articles published under a Creative Commons License, all supplemental material carries the same license as the article it is associated with.
For non-Open Access articles published, all supplemental material carries a non-exclusive license, and permission requests for re-use of supplemental material or any part of supplemental material shall be sent directly to the copyright owner as specified in the copyright notice associated with the article.