
Abstract
Abstract
Various factors affect the process of obtaining stable Arbas cashmere goat embryonic stem cells (ESCs), for example, the difficulty in isolating cells at the appropriate stage of embryonic development, the in vitro culture environment, and passage methods. With the emergence of induced pluripotent stem cell (iPSC) technology, it has become possible to use specific genes to induce somatic cell differentiation in PSCs. We transferred OCT4, SOX2, c-MYC, and KLF4 into Arbas cashmere goat fetal fibroblasts, then induced and cultured them using a drug-inducible system to obtain Arbas goat iPSCs that morphologically resembled mouse iPSCs. After identification, the obtained goat iPSCs expressed ESC markers, had a normal karyotype, could differentiate into embryoid bodies in vitro, and could differentiate into three germ layer cell types and form teratomas in vivo. We used microarray gene expression profile analysis to elucidate the reprogramming process. Our results provide the experimental basis for establishing cashmere goat iPSC lines and for future in-depth studies on molecular mechanism of cashmere goat somatic cell reprogramming.
Introduction
P
Cattle, sheep, goats, and other large animals are important farming and animal husbandry livestock. However, it is very difficult to separate and obtain animal ESC lines and ensure stable passage in vitro while maintaining the characteristics of the stem cells (Behboodi et al., 2011; Pawar et al., 2009; Tian et al., 2006). Little is known about ESC growth characteristics and environment in vitro; several decades of exploration and study have not yielded actual livestock ESC lines. Following research and development efforts, iPSC technology has been successfully applied to monkeys, rats, and pigs (Liao et al., 2009; Liu et al., 2008; Wu et al., 2009). Rat iPSCs were the frst iPSC lines generated before the report of rat ESC lines. This is the proof-of-principle study to demonstrate that the iPSC technique could be applied to species in which ESC lines are difficult to generate. It may also be possible to obtain livestock iPSCs using iPSC technique, and then obtain their ESCs (Liao et al., 2009), which would eventually render establishing large animal ESC lines possible.
Cashmere goats are an important livestock species in our country, and stable cashmere goat ESC lines have not been established. With the rapid development of somatic cell cloning technology, gene transformation to obtain new livestock varieties is a great leap forward for producing new breeds (Damak et al., 1996; Ohkoshi et al., 2003; Wall et al., 1996). The application of ESCs can greatly improve transgenic efficiency, which has been proven in mouse breeding (Capecchi, M.R., 1989; Hanna et al., 2007; Thomas and Capecchi, 1987). In addition, establishing and studying cashmere goat ESC lines can indirectly promote the study of human ESCs, because the evolutionary tree shows that goats are more closely related to humans than mice are (Dattena et al., 2006). However, the absence of cashmere goat ESCs has hindered development in this field. Therefore, iPSCs are another possible means of obtaining goat ESCs (Takahashi and Yamanaka, 2006).
Ren et al. (2011) reported that goat iPSCs (giPSCs) obtained following the transfer of OCT4, SOX2, c-MYC, KLF4, SV40LT, and hTERT into goat primary ear fibroblasts and reprogramming had similar morphology and cell characteristics to mouse ESCs. The authors were unable to generate stable giPSCs using the classic OSMK combination (OCT3/4, SOX2, c-MYC, KLF4). In 2013, Song et al. (2013) generated iPSCs using the OSMK combination, but these were similar to human ESCs. In the present study, we used OSMK, the small chemical molecules valproic acid (VPA) (Huangfu et al., 2008; Teng et al., 2010) and LiCl (Wang et al., 2011), and the organic small molecule vitamin C (Vc) (Esteban et al., 2010) to generate cashmere goat PSCs, described their biological characteristics, and performed whole-genome microarray analysis on the obtained giPSCs. This study not only provides the experimental basis for establishing a cashmere goat PSC culture system, but also understanding of the mechanism of cashmere goat PSC reprogramming at a molecular level, and reference material for further establishment of cashmere goat ESC lines.
Materials and Methods
Cell culture
The Arbas cashmere goat fibroblasts used in this report were obtained from an Arbas cashmere goat fetus at day 40 of gestation. The isolation of goat fetal fibroblasts (GFFs) was done as previously described. Briefly, an explanted goat fetus was dissociated manually and then treated with 0.25% trypsin–EDTA (Gibco). Primary cultures were grown on tissue culture plates coated with 0.1% gelatin (Sigma-Aldrich) until the first passage, after which standard tissue culture plates were used.
Cells were cultured using Dulbecco's Modified Eagle Medium (DMEM; HyClone) supplemented with 10% fetal bovine serum (FBS; HyClone). Upon reaching 90% confluence, GFFs were passaged with 0.25% trypsin–EDTA and infected with reprogramming virus between passages 3 and 5. We maintained 293T cells (American Type Culture Collection, Manassas, VA, USA) in the same medium. The giPSCs were maintained in ESC medium composed of KnockOut DMEM (Gibco-BRL) supplemented with 20% KnockOut Serum Replacement (KSR, Gibco) or FBS (Gibco), 0.1 mM β-mercaptoethanol (Millipore), 1% nonessential amino acids (Gibco), 2 mM GlutaMAX (Gibco), 1 mM VPA (Sigma-Aldrich), 0.05 mM Vc (Sigma-Aldrich), and 1 ng/mL murine leukemia inhibitory factor (mLIF; Millipore). The giPSCs were passaged by enzymatic dissociation using 1 mg/mL collagenase IV (Gibco) and maintained on feeder layers of mouse embryonic fibroblasts (MEFs) that were mitotically inactivated by mitomycin C (Sigma). Permission to handle all animal samples was granted by the Inner Mongolian University Health Science Center Ethical Committee [SCXK (Mongolia) 2002-0001].
Lentivirus production and infection
293T cells were plated at 6×106 cells per 100-mm dish and incubated overnight. The generation and structure of doxycycline (Dox; Sigma-Aldrich)-controlled tetracycline (Tet)-on-inducible lentiviruses expressing mouse-derived Oct4, Sox2, c-Myc, and Klf4 has been described previously (Brambrink et al., 2008; Li et al., 2011; Wernig et al., 2008). Green fluorescent protein (GFP) complementary DNA (cDNA) was cloned into the EcoRI sites of the same vector backbone (FUW-tetO). Expression of reverse Tet transactivator (rtTA) was driven by a constitutively active human ubiquitin C promoter alpha promoter in the FUW lentiviral backbone (FUW-M2rtTA).
To produce lentiviral particles for stable reprogramming of fibroblasts into iPSCs, 293T cells were transfected with a mixture of viral plasmid and packaging constructs expressing the viral packaging functions and vesicular stomatitis virus G (VSV-G) protein using Lipofectamine LTX (Invitrogen). Viral supernatant was harvested at 24 h and 48 h after transfection. A total 30 mL of supernatant was typically harvested per virus. Viral supernatant was filtered through a 0.45-mm syringe filter (Millipore) and loaded into an Amicon Ultra-15 Centrifugal Filter (Millipore) for concentration. The viral supernatant was concentrated approximately 100-fold by centrifugation at 6000 rpm for 20 min at 4°C. Viral concentrates were stored at −80°C; infection was carried out on 35-mm tissue culture plates in 1 mL of medium containing 5 μg/mL Polybrene (Sigma-Aldrich) with 5–10 μL of each viral concentrate. GFFs were infected at a density of 5×104 cells/plate, and the medium was replaced 24 h after infection. Infection was repeated three times, and cells were subsequently dissociated by trypsin and transferred to plates coated with MEF feeder layers. To induce reprogramming, the culture medium was replaced by ESC medium supplemented with 2 mg/mL Dox.
Optimization of culture conditions
To optimize culture conditions, we designed five ESC culture medium types: (1) FBS, KnockOut DMEM, 0.1 mM μ-mercaptoethanol, 1% nonessential amino acids, 2 mM GlutaMAX, 1 ng/ml mLIF, and 2 mg/mL Dox; (2) KSR, KnockOut DMEM, 0.1 mM β-mercaptoethanol, 1% nonessential amino acids, 2 mM GlutaMAX, 1 ng/mL mLIF, and 2 mg/mL Dox; (3) FBS, KnockOut DMEM, 0.1 mM β-mercaptoethanol, 1% nonessential amino acids, 2 mM GlutaMAX, 1 ng/mL mLIF, 0.05 mM Vc, and 2 mg/mL Dox; (4) FBS, KnockOut DMEM, 0.1 mM β-mercaptoethanol, 1% nonessential amino acids, 2 mM GlutaMAX, 1 ng/mL mLIF, 1 mM VPA, and 2 mg/mL Dox (cells were treated with 5 mM LiCl on days 5–12 after virus infection) (Wang et al., 2011); (5) FBS, KnockOut DMEM, 0.1 mM β-mercaptoethanol, 1% nonessential amino acids, 2 mM glutaMAX, 1 ng/mL mLIF, 1 ng/mL mLIF, 1 mM VPA, 0.05 mM Vc, and 2 mg/mL Dox (cells were treated with 5 mM LiCl on days 5–12 after virus infection) (Wang et al., 2011). On day 28 after virus infection, the clones formed by every 5×104 cells in the five culture medium types were counted.
RNA isolation and reverse transcription
Total RNA was purified using an RNeasy Mini Kit (Qiagen) as per the manufacturer's instructions. Approximately 1 mg of total RNA from each sample was used in a RevertAid First Strand cDNA Syntheses Kit (Thermo Scientific). The PCR products were resolved on 2% agarose gels and visualized by ethidium bromide staining. Images were captured using a gel imaging system (Carestream 212 PRO). The primer sequences employed are listed in Table S1 (Supplementary Data are available at www.liebertpub.com/cell/).
Real-time PCR
Total mRNA was isolated using TRIzol (Invitrogen); 1 μg of RNA was used to synthesize cDNA using a PrimeScript RT Reagent Kit (cat. no. DRR037A, Takara) according to the manufacturer's protocol. Real-time PCR was performed using SYBR Premix Ex Taq (Takara) and analyzed with a Stratagene Mx3000P thermocycler. For semiquantitative PCR analysis, the cDNA solution was amplified over 30 cycles at the optimal annealing temperature. The primer sequences employed are listed in Table S2.
Alkaline phosphatase staining and immunocytochemistry
Alkaline phosphatase (AP) staining was performed using an AP kit (BCIP/NBT; Promega) according to the manufacturer's instructions. For immunocytochemistry, cells were fixed with 4% paraformaldehyde for 10 min at room temperature. After washing with phosphate-buffered saline (PBS), cells were treated with PBS containing 10% normal bovine serum albumin (BSA; Sigma-Aldrich). Then, the cells were incubated with 0.1% Triton X-100 for 30 min at room temperature and incubated with primary antibodies at 4°C overnight. The primary antibodies were antibodies against stage-specific embryonic antigen (SSEA)-1 (1:100, ES Cell Marker Sample Kit; Chemicon), SSEA-3 (1:100, Chemicon), SSEA-4 (1:100, Chemicon), Tra-1-60 (1:100, Chemicon), Tra-1-81 (1:100, Chemicon), Nanog (1:100; Abcam), Oct4 (1:100, Chemicon), Sox2 (1:100, Chemicon), βIII tubulin (1:100, Abcam), desmin (1:100, Abcam), and cytokeratin (1:100, Abcam). Normal mouse or rabbit serum was used as the negative control. Antigen localization was visualized with anti-rabbit or anti-mouse immunoglobulin G (IgG) secondary antibodies conjugated with fluorescein (Santa Cruz Biotechnology). Nuclei were counterstained with diaminophenylindole.
Transcriptional profiling by microarray
An RNeasy Mini Kit (Qiagen) was used to extract RNA from giPSCs from passage 7 GFFs and from passage 1 inner cell mass (ICM)-like ESCs from 20-day goat blastocysts. A WT Expression Kit (4411973; Ambion, Austin, TX, USA) was used to prepare RNA samples for Agilent whole-genome microarray analysis (Santa Clara, CA, USA); RNA was labeled using a One-Color QuickAmp Labeling Kit (cat. no. 5190-0442, Agilent). The fragmented and labeled samples were hybridized to Agilent Goat Whole Genome Oligo Microarrays, which are based on the National Center for Biotechnology Information (NCBI) BioProject PRJNA217901. The goat array contains 29,496 probe sets representing >22,981 transcripts. The data were normalized by MAS5. Differentially expressed genes were processed using Cluster 3.0; the Euclidean distance was used as the cluster method for heat map generation. The two-fold change in gene expression was used as the cutoff level for data analysis.
In vitro differentiation
Spontaneous differentiation of giPSCs through embryoid body (EB) formation was carried out as previously described (Li et al., 2011; Takahashi and Yamanaka, 2006). Briefly, giPSCs were cultured in ESC medium without mLIF and Dox in non–tissue culture-treated plates. After 8 days in suspension culture, EBs were transferred to gelatin-coated plates and cultured in differentiation medium for another 8 days.
Teratoma formation
The giPSCs were harvested by collagenase IV treatment, suspended in PBS, and injected subcutaneouslyinto the dorsal flanks of severe combined immunodeficient (SCID) mice. Eight weeks later, the resultant tumors were explanted, fixed in 4% paraformaldehyde, embedded in paraffin, and examined histologically using Hematoxylin & Eosin staining.
Karyotyping
The giPSCs were prepared for karyotype analysis by 5 h of incubation in medium containing 0.1 mg/mL colcemid. Cells were trypsinized, resuspended in 0.075 M KCl, incubated at 37°C for 30 min, and fixed in 3:1 methanol:acetic acid at room temperature for 5 min. Centrifugation and fixing were repeated thrice.
Results
Production of giPSCs and AP staining
Figure 1A depicts the giPSC induction timetable. Infected cells (day 4) were plated in a MEF feeder layer–coated six-well plate. Cell morphology began to change at about day 7 of infection, and there was faster proliferation. On day 12, the cells were digested into single-cell suspensions and plated onto another feeder layer. On day 20, ESC-like clones could be observed. They were elliptic and had distinct margins, similar to mouse ESCs. The clones increased significantly after 25 days. On day 28 (Fig. 1B, a–f), some clones were selected into 96-well plates, and amplification cultures, namely the giPSCs1, giPSCs2, and giPSCs3 lines, were obtained. The nuclear-to-cytoplasmic ratio was high, proliferation speed was low, and the cells could be passaged again after about 10 days. Following type IV collagenase digestion, giPSCs have already been stably passaged for more than 15 generations (more than 100 days). The average doubling time of giPSCs was about 22.14 h and the average plating effciency was 25.4±0.17%.

Morphological changes of GFFs under going reprogramming to iPSCs. (
The remaining clones were stained with AP, and clone formation efficiency was calculated. Approximately 80% of the clones were AP positive (Fig. 1B, i). The medium was changed to no-Dox medium and the cultures maintained; 94% of the initially AP positive clones became AP negative, losing their original form and differentiating rapidly (Fig. 1B, h).
Optimization of culture medium and addition of small molecules
FBS- or KSR-containing induction medium was used to cultivate virus-infected cells, and AP-positive clones were calculated in 5×104 cells after 28 days. Cells in FBS- and KSR-containing induction medium formed an average of 10.3±2.1 and 4.7±1.2 AP-positive clones, respectively. There were significantly more AP-positive clones in the FBS-containing medium than in the KSR-containing medium (Fig. 2A).

AP-positive clones were calculated after optimization of culture conditions and immunofluorescence staining of giPSCs. (
Small molecular compounds were added to FBS-containing induction medium containing large numbers of AP-positive clones, the cells were cultured, and AP-positive clones were calculated in every 5×104 cells. Cells cultured in FBS and Vc medium, FBS, VPA, and LiCl medium, or FBS, Vc, VPA, and LiCl medium formed an average 11.5±1.3, 16.8±2.1, and 22.9±0.75 AP-positive clones, respectively (Fig. 2B).
Pluripotent marker gene expression in giPSCs
Immunofluorescence was used to detect specific labeled proteins in the ESCs of fifth-generation giPSC clones, which were positive for the ESC-specific marker proteins Oct4, Sox2, Nanog, SSEA-1, Tra-1-60, and Tra-1-81, and negative for SSEA-3 and SSEA-4 (Fig. 2C).
Reverse transcription PCR and real-time quantitative detection of giPSC pluripotent marker genes
Fourth-generation giPSCs clones were harvested, and the total RNA was extracted for reverse transcription PCR (RT-PCR). With Dox, there was high expression of the exogenous genes OCT4, SOX2, KLF4, and c-MYC (Fig. 3A), which continued for >15 generations, whereas their expression in clones in the no-Dox medium decreased rapidly (Fig. 3B). The giPSCs expressed endogenous OCT4, SOX2, NANOG, KLF4, LIN28, REX1, PODXL, DNMT3B, and other stem cell–related genes (Fig. 3C).

RT-PCR of three giPSC lines compared with GFFs. (
GFFs were used as the control, and real-time quantitative PCR was conducted to detect OCT4, SOX2, NANOG, KLF4, and LIN28 expression. OCT4, NANOG, and KLF4 were highly expressed in the giPSCs, but SOX2 and LIN28 expression was low (Fig. 3D). By contrast, OCT4, SOX2, NANOG, KLF4, and LIN28 expression was almost absent in the control group.
Whole-genome expression profile microarray analysis of giPSCs
Seventh-generation giPSCs were harvested for whole-genome expression profile microarray analysis. Heat maps of selected data that were generated from Agilent gene chip analysis are shown in Figure 4A. In addition, the log2-fold changes in expression levels of pluripotency genes in giPSCs compared with ICM-like ESCs and GFFs are provided (Fig. 4B). Theirs was obviously different from that of the GFFs, and significant differences persisted when compared with the ICM-like ESCs. We determined the relative expression of PSC-related genes, including OCT4, SOX2, NANOG, DNMT3B, KLF5, and PODXL. There was obviously higher expression of the above genes in the giPSCs compared to the GFFs, but overall expression was lower than that in ICM-like ESCs.
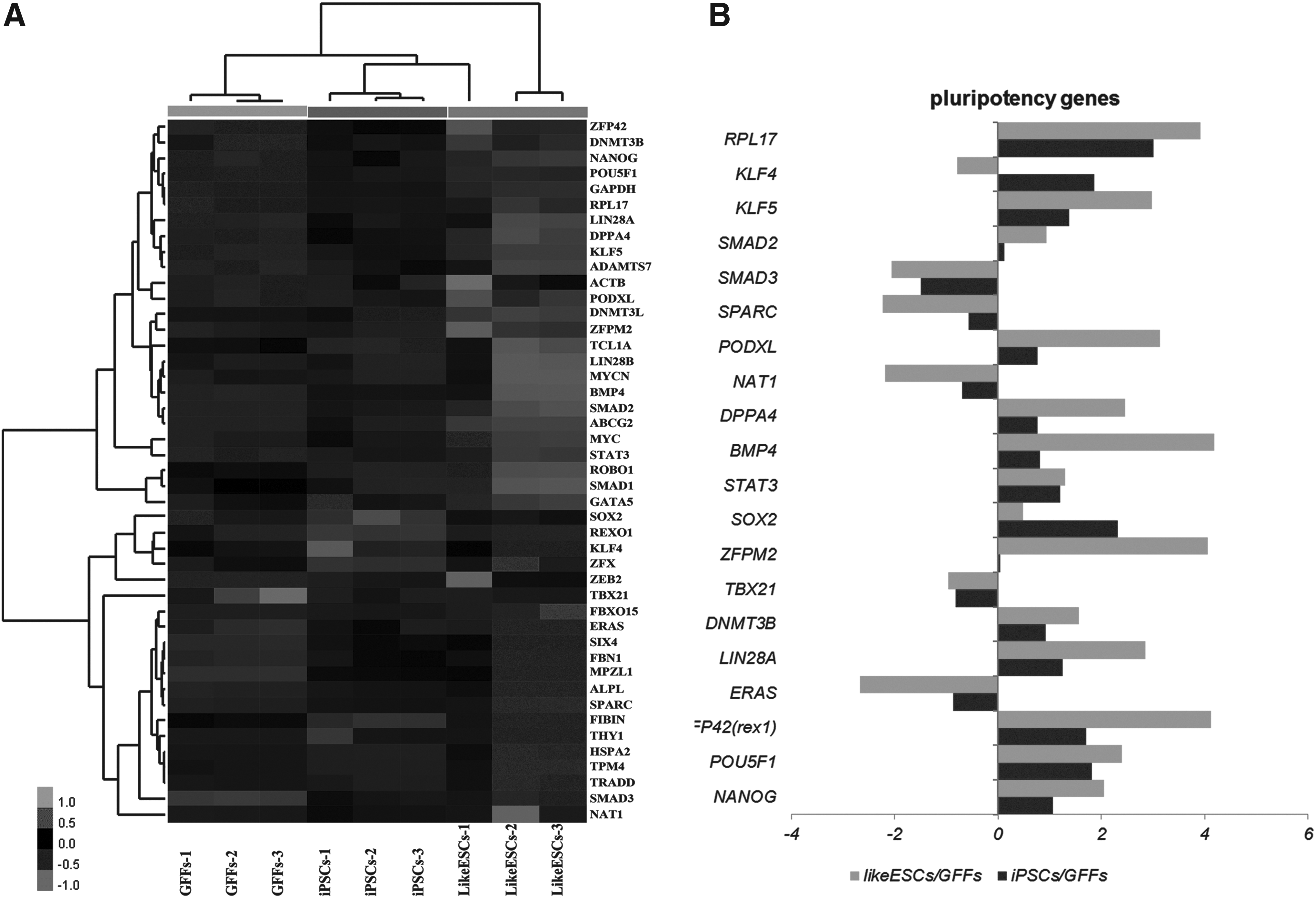
Gene expression profiling of giPSCs using Agilent microarrays. (
In vitro and in vivo differentiation ability and karyotype analysis of giPSCs
Culture medium without mLIF, Vc, and VPA was used to culture passage 7 giPSCs, and EB clusters were observed after 8 days (Fig. 5A). The EBs were transferred to gelatin-coated plates and cultured for an additional 8 days, and immunofluorescence was used to detect the ectodermal, mesodermal, and endodermal markers βIII tubulin, desmin, and cytokeratin, respectively, for which immunofluorescence was all positive (Fig. 5B–D), indicating differentiation into the three germ layer cell types.

The giPSCs differentiated into derivatives of all three germ layers in vitro and in vivo. The giPSCs formed EBs (
Passage 6 giPSCs were injected into the skin over the left side of the spine in nonobese SCID mice; distinct protuberances appeared after 8 weeks. Tissue sections were stained, and the endoderm (glandular-like tissue), mesoderm (muscle), and ectoderm (neural tissue) layers were detected (Fig. 5E–G), showing that the teratomas contained differentiation into three germ layer cell types. Karyotype analysis of passage 15 giPSCs determined that they had normal chromosome numbers, i.e., 58+XY (Fig. 5H).
Discussion
Compared to mice (Evans and Kaufman, 1981), hamsters (Doetschman et al., 1988), rhesus monkeys (Liu et al., 2008), and rats (Liao et al., 2009), establishing stable ESC lines in large livestock animals is difficult. Factors such as selecting the appropriate stage of embryo development to isolate the ICM, the suboptimal culture conditions, and different passage methods in vitro greatly affect the establishment of large animal ESC lines. However, with iPSC technology, it has become possible to induce terminally differentiated cells from PSCs using specific factors, such as how swine and canine iPSCs are established (Esteban et al., 2009; Ezashi et al., 2009). At present, the establishment of goat stable ESC lines has not been reported. In 2011, Ren et al. (2011) successfully established giPSCs. However, they transferred six genes, reducing the direct application potential of the cell lines, and the stem cell purity of the giPSCs has been questioned. In this experiment, we used only the classic OSMK with small molecules to obtain a giPSC line. It provides a research platform for establishing an authentic ESC line and provides the basis and reference for discovering the molecular mechanism of ESCs.
Morphologically, the giPSCs were similar to mouse ESCs, being dense, round clones with distinct margins and high refractive index. The giPSCs not only expressed ESC marker genes, they also could differentiate into the three germ layer cell types in vitro. However, the giPSCs obtained by Song et al. (2013) were more similar to human ESCs. The exogenous gene expression involved the use of a drug-induced, Dox-dependent Tet-on-inducible system (Wu et al., 2009); following activation, expression of the endogenous genes inhibited exogenous gene expression. Cell reprogramming time is comparatively long, and clones are observed only after about 20 days of subculture following virus infection (Bao et al., 2011). Reprogramming efficiency is determined when 1×104 fibroblasts can form seven to eight AP-positive clones. The expanded cultures contained specific cell differentiation. These results show that reprogramming goat fibroblasts is far more difficult than reprogramming fibroblasts from other species. However, exogenous gene expression decreased in the absence of Dox; the giPSCs could not maintain their original morphology, and the cells differentiated rapidly. This proves that the existing culture environment was insufficient for maintaining the pluripotency of the giPSCs we established under no-Dox conditions (Bao et al., 2011; Li et al., 2011; Ren et al., 2011).
We found that giPSC induction was more effective and complete in FBS-containing medium compared to KSR-containing medium. These results show that some components in the FBS extracellular matrix play an important role in giPSC generation, whereas KSR does not contain those active ingredients.
Recently, small molecules that can enhance the generation of iPSCs or compensate the requirement of certain oncogenic factors will be highly valuable. They not only improve reprogramming efficiency, but also aid in blocking the potential mechanism of differentiation. Numerous small chemical molecules promote reprogramming (Li and Ding, 2010); for example, Vc effectively promotes cell reprogramming (Esteban et al., 2010) and delays cell aging. In the present study, we used several small molecules—the epigenetic modification molecules VPA (Huangfu et al., 2008; Teng et al., 2010) and the inorganic compound LiCl (Wang et al., 2011). We found that the small molecules VPA, LiCl, and Vc effectively promoted cell reprogramming and greatly improved the giPSC culture environment. However, this was also insufficient for maintaining the stem cell characteristics of the giPSCs when Dox was removed.
In this study, we used goat whole-genome expression microarray for the first time. Our results are in accord with the microarray analysis results, differing slightly only in terms of the expression of individual genes. The microarray analysis results involved differential expression, which showed that total giPSC gene expression was significantly different compared to that of GFFs and ICM-like ESCs. The expression of many giPSC genes was lower compared to that of the ESCs. It also shows that there is a discrepancy between giPSCs and actual ESCs, which demonstrates that giPSC reprogramming is still insufficient for completely or thoroughly maintaining their stem cell characteristics under existing culture conditions. This may have been due to the immaturity of the culture environment, where the expression of stem cell–associated genes in giPSCs was slightly lower than that in the ESCs. However, giPSC and ICM-like ESC SOX2 expression was both very low, which quantitative PCR also demonstrated. This finding is consistent with the established practices in stem cell marker gene expression (Buganim et al., 2014; Esteban et al., 2010). The classic OSMK combination is not ideal for inducing giPSCs. The microarray analysis showed that maintaining iPSC stemness is largely dependent on exogenous gene expression; compared with mouse and human iPSCs, there was an obvious difference between giPSC-derived cells and actual goat ESCs.
An important proof for the establishment of ungulate pluripotent stem cell lines is demonstration of pluripotency either by differentiation into defined cell types in vitro or by teratoma formation in vivo. These proofs have been common practice with both mouse and primate ESC and iPSC lines (Evans and Kaufman, 1981; Li et al., 2011; Takahashi and Yamanaka, 2006). In the present study, giPSCs have differentiated into EBs with the three germ layer cell types in vitro and formed teratomas in vivo. However, teratoma formation from giPSCs is more difficult than that of mouse iPSCs. Teratoma formation is slow and only occurs 8 weeks after injection in immunodeficient mice. In the present experiment, giPSCs formed three germ layer EBs and teratomas. However, these giPSCs should be injected into developing embryos in future experiments to obtain a chimera and further prove that these cells have ESC-like cell characteristics.
This study only transferred four factors (Oct4, Sox2, c-Myc, Klf4) into Arbas cashmere GFFs; using a drug-inducible system for the induction culture, we obtained giPSCs that were morphologically similar to mouse ESCs. The giPSCs expressed ESC markers, maintained a normal karyotype, differentiated into EBs with the three germ layer cell types in vitro, and formed teratomas in vivo. The coordinated application of chemical small molecules compensated somewhat for the inadequate culture environment. Whole-genome expression profile microarray analysis further identified the reprogramming conditions and verified the experimental results. Our results provide an experimental basis for the establishment of cashmere goat iPSC lines and for future in-depth studies of the molecular mechanism of cashmere goat somatic cell reprogramming.
Footnotes
Acknowledgment
This work was supported by National Natural Science Foundation of China (no. 31101698), Inner Mongolia Natural Science Foundation (no. 2011JQ03).
Author Disclosure Statement
The authors declare that no conflicting financial interests exist.
References
Supplementary Material
Please find the following supplemental material available below.
For Open Access articles published under a Creative Commons License, all supplemental material carries the same license as the article it is associated with.
For non-Open Access articles published, all supplemental material carries a non-exclusive license, and permission requests for re-use of supplemental material or any part of supplemental material shall be sent directly to the copyright owner as specified in the copyright notice associated with the article.