
Abstract
Abstract
Cryopreservation of spermatogonial stem cells (SSCs) is an applicable method for young males seeking fertility preservation before starting a treatment. It increases reactive oxygen species (ROS) formation and oxidative stress, which damages cellular structures. In this study, we added two antioxidants, catalase and α-tocopherol (α-TCP), to the basic freezing medium to evaluate their effects on the efficiency of SSCs. SSCs were isolated from testes of 3- to 6-day-old male mice using enzymatic digestion. The enrichment of isolated cells was evaluated by flow cytometry and Stra8 antibody. Catalase (40 μg/mL), or α-TCP (200 μg/mL) was added to the basic freezing medium. The cell viability was evaluated by the methylthiazoltetrazolium (MTT) assay. After thawing, cells were cultured for 1 month, and the expression pattern of specific genes of SSCs and the ability of the cells to restore spermatogenesis were used to determine the efficiency of the cryopreservation method. The survival rate of the frozen cells in the presence of catalase or α-TCP was significantly higher than the control group (p < 0.05). The number of colonies and their diameter measured after 1 month were significantly higher in the antioxidant groups than in the control group (p < 0.05). Gene expression and resumption of spermatogenesis also followed the same pattern. Thus, adding antioxidants to the basic freezing medium can be helpful in increasing the quality and viability of SSCs after cryopreservation. This new approach to stem cells cryopreservation can also be a promising strategy for fertility preservation in patients who suffer from malignancy.
Introduction
S
Reduction of germ cell loss is an alternative strategy for preserving the fertility of patients undergoing chemotherapy and irradiation treatments (Shetty et al., 2013). Other strategies that have been used for the preservation of SSCs include long-term culture strategy, SSCs cryopreservation, and transplantation. Izadyar et al. showed that it is not possible to preserve pure populations of SSCs for a long time in a culture system (Izadyar et al., 2002). However, the combination of culture approach with cryopreservation has been found to be a suitable approach for long-term preservation of SSCs. By increasing the number of SSCs, the transplantation technique can then be used to restore spermatogenesis in infertile men (Brinster and Zimmerman, 1994).
Several studies have also demonstrated that the slow-freezing method is an applicable long-term preservation method for SSCs preservation, and fertility can be restored in infertile mice following transplantation of cryopreserved SSCs (Wu et al., 2012). Methods used for cryopreservation of SSCs are similar to those used for somatic cells; however, there are few studies on the evaluation of the effects of cryopreservation on SSCs (Lee et al., 2013). It is well established that the cryopreservation procedure increases reactive oxygen species (ROS) formation and oxidative stress that can damage cellular structures such as membranes, structural proteins, enzymes, and nucleic acids (Bilodeau et al., 2002). In addition, ROS can cause damage in vital tissues and cell functions and endangers cell viability (Bilodeau et al., 2000).
To prevent oxidative stress damage to cellular structures, it is necessary to have a complex network of antioxidants and enzymes in organisms. Antioxidants can combat free radicals by terminating the reaction chain in mitochondrial membranes and thus can protect molecules from oxidative damage (Kothari et al., 2010). Both kinds of antioxidants (enzymatic and nonenzymatic types) can neutralize excess ROS and prevent them from causing damage to cellular structures (Fernández‐Santos et al., 2007).
α-Tocopherol (α-TCP) is an antioxidant belonging to the vitamin E family, a family including α-, β-, γ-, and δ-TCP, that possesses biological properties (Azzi, 2007; Singh et al., 2006). Studies have shown that α-TCP has significant effects on cell signaling, regulation of gene expression, inflammation, cell proliferation, and apoptosis (Lemaire‐Ewing et al. 2010). Effects of vitamin E have been explained by its antioxidant functions, including the scavenging of ROS and reactive nitrogen species (Zingg 2007). α-TCP is a biological antioxidant that decreases ROS levels and oxidative stress markers in living organisms (Winterbone et al., 2007). Several studies have shown that the application of α-TCP prior to irradiation increases hematopoietic stem cell survival in mice (Roy et al. 1982). Catalase (CAT), the other antioxidant used in this study, is an enzymatic antioxidant located in a cell organelle called the peroxisome that rapidly promotes the conversion of hydrogen peroxide (H2O2) to water and molecular oxygen (Valko et al., 2006).
In this study, we tried to improve the conditions for cryopreservation of SSCs, after which further assessment studies were made. The effects of α-TCP and CAT treatment on the cryopreservation, proliferation, and transplantation of SSCs were evaluated in this study. Our findings showed a novel and innovative cryomedium for SSCs, using the antioxidants CAT and α-TCP in the cryomedium, that prevents the release of free radicals during SSC cryopreservation and enhances their survival rate after thawing and transplantation.
Materials and Methods
Animals
Three- to 6-day-old male National Medical Research Institute (NMRI) mice (day of birth is 0) were maintained under standard laboratory conditions. All experiments were approved by the Ethics Committee of Tehran University of Medical Sciences and performed in accordance with the University guidelines.
Isolation of SSCs
Two-step enzymatic digestions were used to isolate SSCs from testis tissue (five mice at a time in each group). Testes of NMRI mice were collected and washed in phosphate-buffered saline (PBS; Sigma, Munich, Germany). The tunica albuginea was removed, and the testes were minced into small pieces and transferred to the digestion medium containing collagenase type IV (1 mg/mL; Sigma), DNase (10 μg/mL; Sigma), and hyaluronidase (0.5 mg/mL; Sigma) in minimum essential medium alpha (MEMα; Sigma) for 20 min, with shaking at 37°C in 5% CO2 until the tubules were separated. The cells were then centrifuged at 1500 × g for 5 min, after which they were washed twice with PBS. The second step of digestion was performed for testis cells suspension with the same enzymes used above (15 min). The digested cells were then washed with PBS (Guan et al. 2009).
Cryopreservation
After cell isolation, three different freezing media were slowly added to the cell suspension: (1) Control group: The basic freezing medium consisted of dimethyl sulfoxide (DMSO; 1.4 M, Sigma), 10% fetal bovine serum (FBS; Sigma), and MEM-α (Sigma) (control group) (Izadyar et al. 2002). (2) CAT group: 40 μg/mL CAT (Sigma) was added to the basic freezing medium. (3) α-TCP group: 200 μg/L α-TCP (Sigma) was added to the basic freezing medium. Cryovials were placed into a Nalgene CryoBox (cat. no. Z359017, Sigma) and stored in a −80°C freezer for at least 1 day. After overnight storage, cryovials containing frozen cells were immersed into liquid nitrogen for at least 1week.
Thawing procedure
After removal from liquid nitrogen, samples were maintained at room temperature for 30 sec, and then in a water bath at 37°C for 2 min. The cryovial contents were transferred to a tube with prewarmed medium (MEM-α + 10% FBS). The cells were washed two times with medium and centrifuged at 1200 × g for 5 min (Izadyar et al., 2002). After removing the supernatant solution, the cell pellet was subjected to viability assessment, purity percentage, and gene expression by real-time polymerase chain reaction (real time PCR), cell culture, and transplantation.
Cell viability
Cell viability was measured by the methylthiazoltetrazolium (MTT; Sigma, Germany) assay before and after cryopreservation. MEM (400 μ)L) and MTT (40 μL) were added to each well (400 cells for each condition) and incubated for 4 h at 37°C. Subsequently, medium was replaced by 400 μL of DMSO. The cells were kept at room temperature for 30 min, and the optical density (OD) at 540 nm was measured afterward using a microplate reader.
Enrichment percentage assessment
Flow cytometry was performed to assess the cellular enrichment percentage before and after cryopreservation and after SSC culture applying standard procedures. A total of 106 cells were centrifuged at 1200 × g for 7 min, and the cell pellet was resuspended in 100 μL of PBS/FBS and was fixed with Triton X-100. To identify Stra8-positive cells, 10 μL of primary antibody (anti-Stra8 antibody, cat. no. ab49602, Abcam, Cambridge, UK) was added to the cell suspension (1 h, at room temperature). After 1 h, cells were washed in 1 mL of PBS/FBS, and 10 μL of secondary antibody (donkey anti-rabbit, cat. no. ab6798, Abcam) conjugated with fluorescein isothiocyanate (FITC) was added (1 h, 4°C). Cells used as controls were not treated with antibodies. Cells were kept in the dark on ice until analysis by flow cytometry (Baazm et al., 2013).
SSCs culture
In all groups, SSCs were cultured for 1 month after cryopreservation (density of 2 × 105 cells/cm2). Basic culture medium contained MEMα, 10% FBS, 1× nonessential amino acids (Invitrogen, Carlsbad, CA, USA), 0.1 mM 2-mercaptoethanol (Sigma), 103U/mL human recombinant leukemia inhibitory factor (LIF; B&D, Franklin Lakes, NJ, USA), 100 U/mL penicillin, and 100 μg/mL streptomycin (both from Sigma). To each culture dish, we added 10 μg/mL glial cell line–derived neurotrophic factor (GDNF; R&D Systems, Minneapolis, MN, USA). The culture medium was changed every 3 days. All culture dishes were maintained at 32°C in an atmosphere humidified with 5% CO2, and cells were passaged every 7–10 days during the period of culture (four passages).
Colony assay
The diameter and number of colonies were measured at the end of first and fourth weeks (7 and 30 days after seeding in all groups). Colony formation occurred earlier (day 4) in the antioxidant groups than in the control (day 7). We counted all colonies in each 30-mm dish in each category (this was repeated three times). Colony diameters were measured using an ocular grid on an inverted microscope. Captured images were processed by imageJ software (Koruji et al., 2007).
Real-time polymerase chain reaction
Before and after 1 month of culture, we examined the expression levels of the apoptosis regulator BAX (Bax, pro-apoptotic), B cell lymphoma 2 (Bcl2, antiapoptotic), tyrosine protein kinase Kit (c-kit, differentiated gene), promyelocytic leukemia zinc finger protein (Plzf, undifferentiated gene), and DNA-binding protein inhibitor ID-4 (ID4, undifferentiated gene) for the evaluation of optimal conditions for SSCs cryopreservation,. The primer sequences are shown in Table 1. Total RNA was extracted by the RNeasy Kit (Ready Mini Kit, Qiagen, Valencia, CA, USA), according to the manufacturer's instructions. cDNA synthesis was performed using a reverse transcription kit (Transcriptor First Strand cDNA Synthesis Kit, Roche, Branchburg, NJ, USA) with 1 μg of total RNA, according to the manufacturer's instructions. Real-time PCR was performed with 40 reaction amplification cycles. 7500 Fast Real-Time PCR with SYBR Green detection (Applied Bioscience, Carlsbad, CA, USA) was used for the analysis. Melt curve analysis was performed after each run to check for the presence of nonspecific PCR products and primer dimers. All samples were normalized against glyceraldehyde-3-phosphate dehydrogenase (GAPDH) (internal control) using the comparative CT method (ΔΔCT).
F, forward; R, reverse.
Transplantation
After 1 month of culture, SSCs from all groups were transplanted into one of the testes of a recipient mouse. Before transplantation, 40 mg/kg busulfan (Sigma) was injected intraperitoneally into 6-week-old male mice. After 1 month, 10 μL of the donor cell suspension (107cells/mL) was injected into the seminiferous tubules of the recipient testis through the efferent duct. The other testis was used as an internal control (Brinster and Avarbock, 1994). Before the injection, SSCs were labeled with the red fluorescent cell linker di-alkyl indocarbocyanine (DII; Sigma), according to the manufacturer's protocol. Immediately after trypsinization, the cells were washed with medium (without serum) and centrifuged at 1500 × g for 5 min; the pellet was resuspended in medium. DII (24–32 × 106 mol) was prepared and immediately added to the cells, which were incubated for 8–10 min.
After labeling, serum was added and the cells were washed with medium. SSCs were counted, and Trypan Blue was used for monitoring the successful entry of cells into the seminiferous tubules. After 2 months, the recipient mice were killed and the testes were dissected for detection of labeled SSCs. Samples were fixed in 4% paraformaldehyde and embedded in paraffin (Sigma, Germany) for routine histologic processing and Hematoxylin & Eosin (H&E) staining (Honaramooz et al., 2002, 2003). For each testis, 50 sections were prepared and 100 tubules were evaluated in each section (Abbasi et al., 2015).
Statistical analysis
Results were expressed as mean ± standard deviation (SD). The statistical significance between the mean values was determined by one-way analysis of variance (ANOVA) followed by a Tukey post hoc test with p ≤ 0.05 as the statistically significant criterion. A total of three independent experiments were performed.
Results
Enrichment of SSCs
After enzymatic digestion, flow cytometry analysis showed that 43.54% of all cells expressed Stra8. Results obtained after freezing showed that the enrichment of SSCs in the CAT (71.45%) (p < 0.05) and α-TCP (61.54%) (p ≤ 0.05) groups was significantly higher than in the control group (52.63%) (p ≤ 0.05) (Fig. 1A–D). After 1 month of culture, analysis showed that the enrichment of SSCs in the CAT (81.34%) (p ≤ 0.05) and α-TCP (71.56%) (p ≤ 0.05) groups was significantly higher than in the control group (60.34%) (p ≤ 0.05) (Fig. 1E–G).
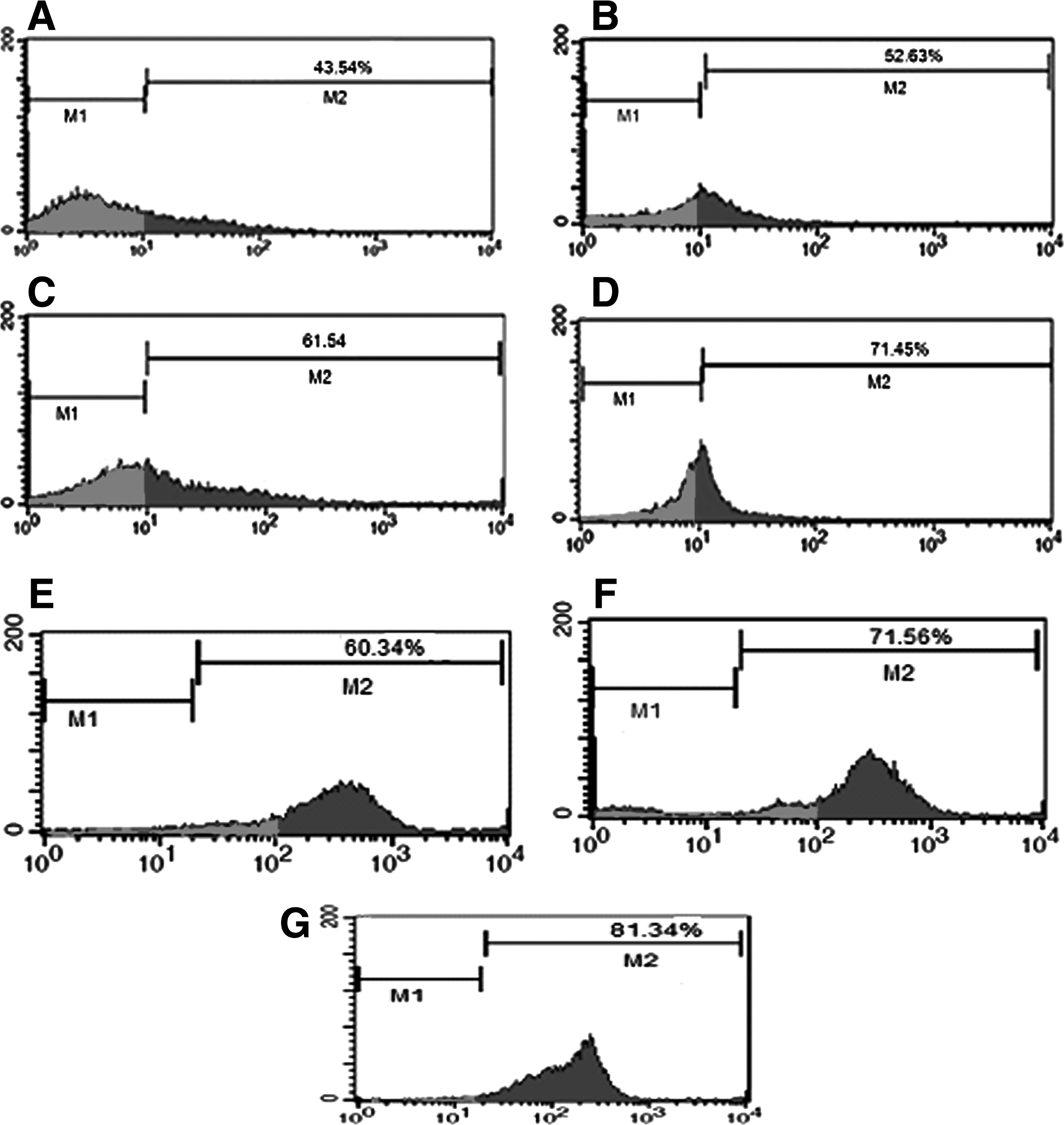
Flow cytometry analysis for the detection of the Stra8 marker in fresh cells, treated groups after cryopreservation, and culture. (
Effect of cryopreservation on SSC viability
MTT assay results showed that the majority of the cells (89.85 ± 6%) were viable immediately after enzymatic digestion. After 1 week of cryopreservation, the survival rate of cryopreserved cells in the control group (70.65 ± 6%) was significantly lower than in fresh cells (89.85 ± 6%) (p ≤ 0.0001). Furthermore, cell viability was significantly (p ≤ 0.0001) increased in 40 μL of CAT (81.45 ± 5%) and 200 μL of α-TCP (79.31 ± 4%) compared with the control group (70.65 ± 6%) (Fig. 2).
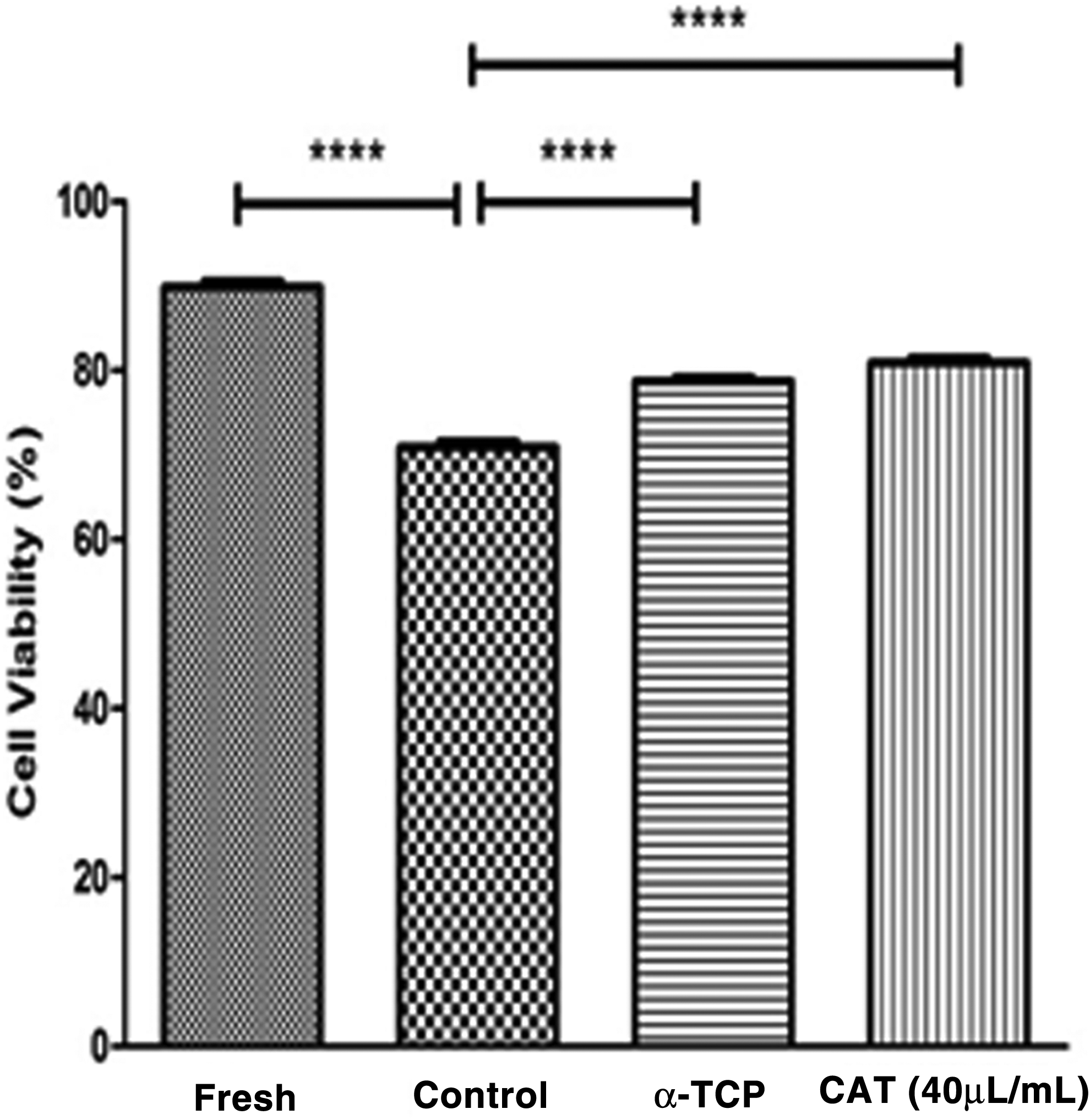
MTT assay analysis of viability in different groups. Results indicated that the survival rate of cryopreserved cells in the control group was significantly lower than in fresh cells. Antioxidant groups have an increased viability in compared with control group. Data show means ± SD; (****) p ≤ 0.0001.
SSCs culture
After cryopreservation, SSCs from all groups were cultured for 1 month. On the first day of culture, SSCs were single and attached to the dish, but colonies formed after some days. The colonies appeared significantly earlier (on day 4) in the antioxidant groups compared with colony formation time in the control group (on day 7 or after seeding) (Fig. 3). The number and diameter of colonies were compared in all groups. Colony assay indicated that the number of colonies and their diameters in α-TCP (20.5 ± 3.1 and 290.3 ± 50μ) and CAT (24.7 ± 5.1 and 350.5 ± 50 μm) groups were significantly higher than in the control group (15.1 ± 2.4 and 234.27 ± 50 μm) (p ≤ 0.001) (Fig. 3). The results thus show that CAT had a wider effect on diameter and frequency of colonies (Fig. 4A, B).
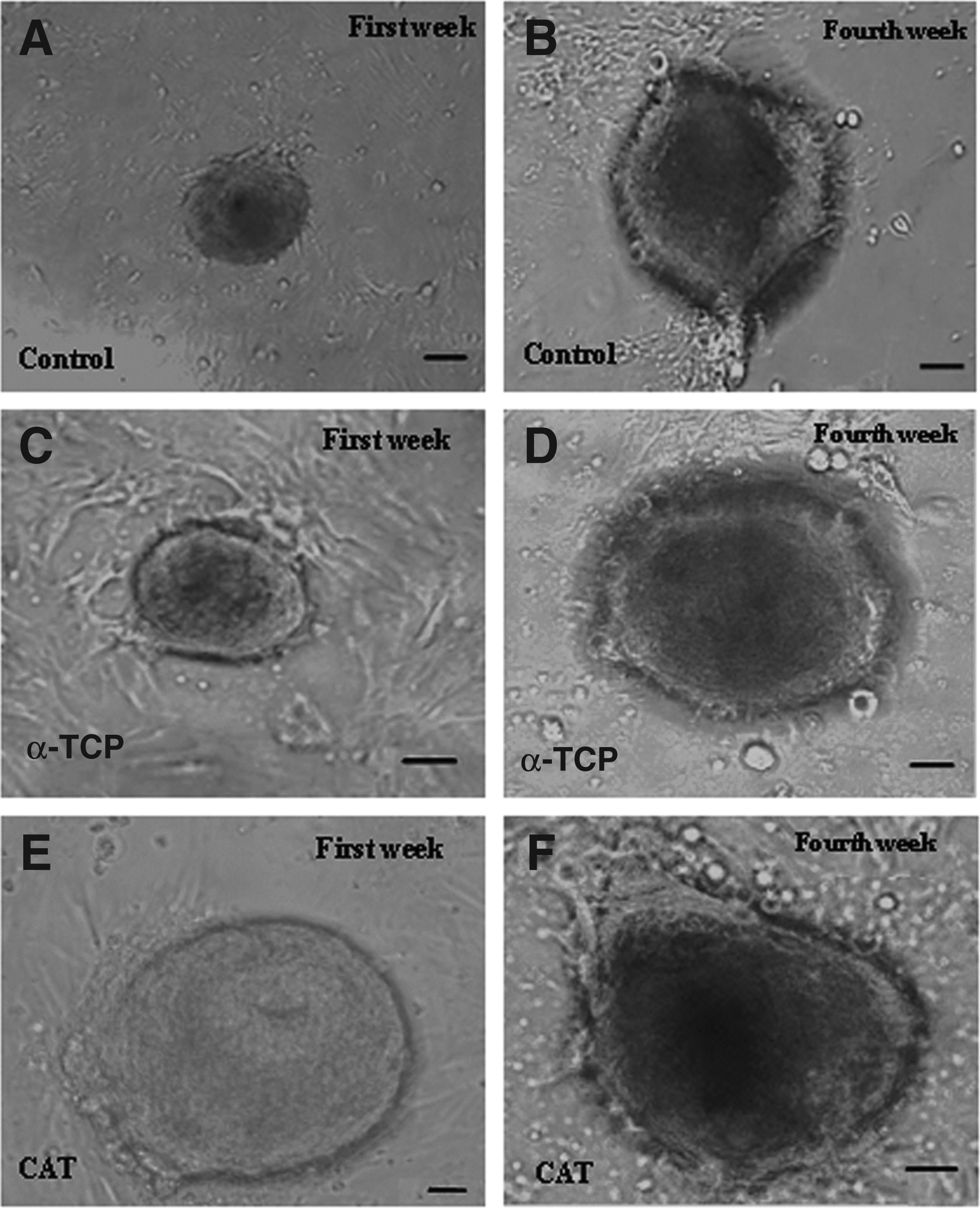
Morphology of SSCs derived from 3- to 6-day-old male mice. The sizes of colonies are shown at the first and fourth weeks of culture. After cultivation of SSCs, diameter and number of colonies increased in all groups, especially in the CAT group. (
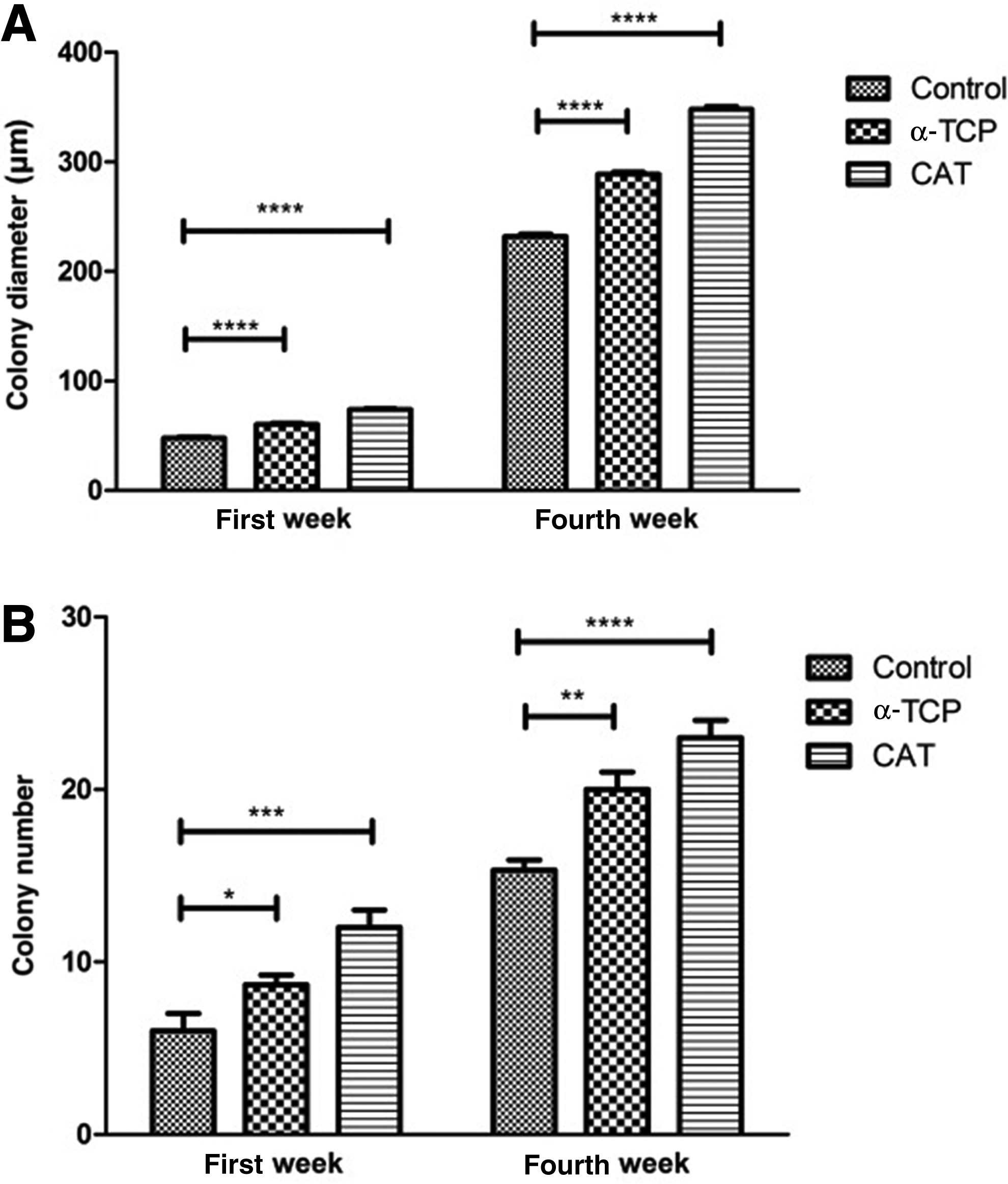
Comparison of colony diameters (
Gene expression
After cryopreservation, results indicated that the level of Bax and Bcl2 expression in the CAT and α-TCP groups was significantly (p ≤ 0.0001) lower and more than in the control group, respectively. ID4 and Plzf expression in the antioxidant groups was increased (p ≤ 0.05) compared with the control group, whereas c-kit gene had a decreased level of expression in the antioxidant groups compared with the control group. After culture, the level of Bax expression in the CAT and α-TCP groups was significantly (p ≤ 0.0001) lower than in the control group; however, the level of Bcl2 expression was significantly (p ≤ 0.0001) higher than in the control group. Genes related to undifferentiated state, ID4 and Plzf, in the antioxidant groups had an increased level of expression (p ≤ 0.05) compared with the control group, whereas the c-kit gene had a decreased but not significantly lower expressed level in the antioxidant groups compared with the control group. On the basis of these results, it appears that treatment with CAT and α-TCP had a higher effect on gene expression levels, especially with CAT (Fig. 5).
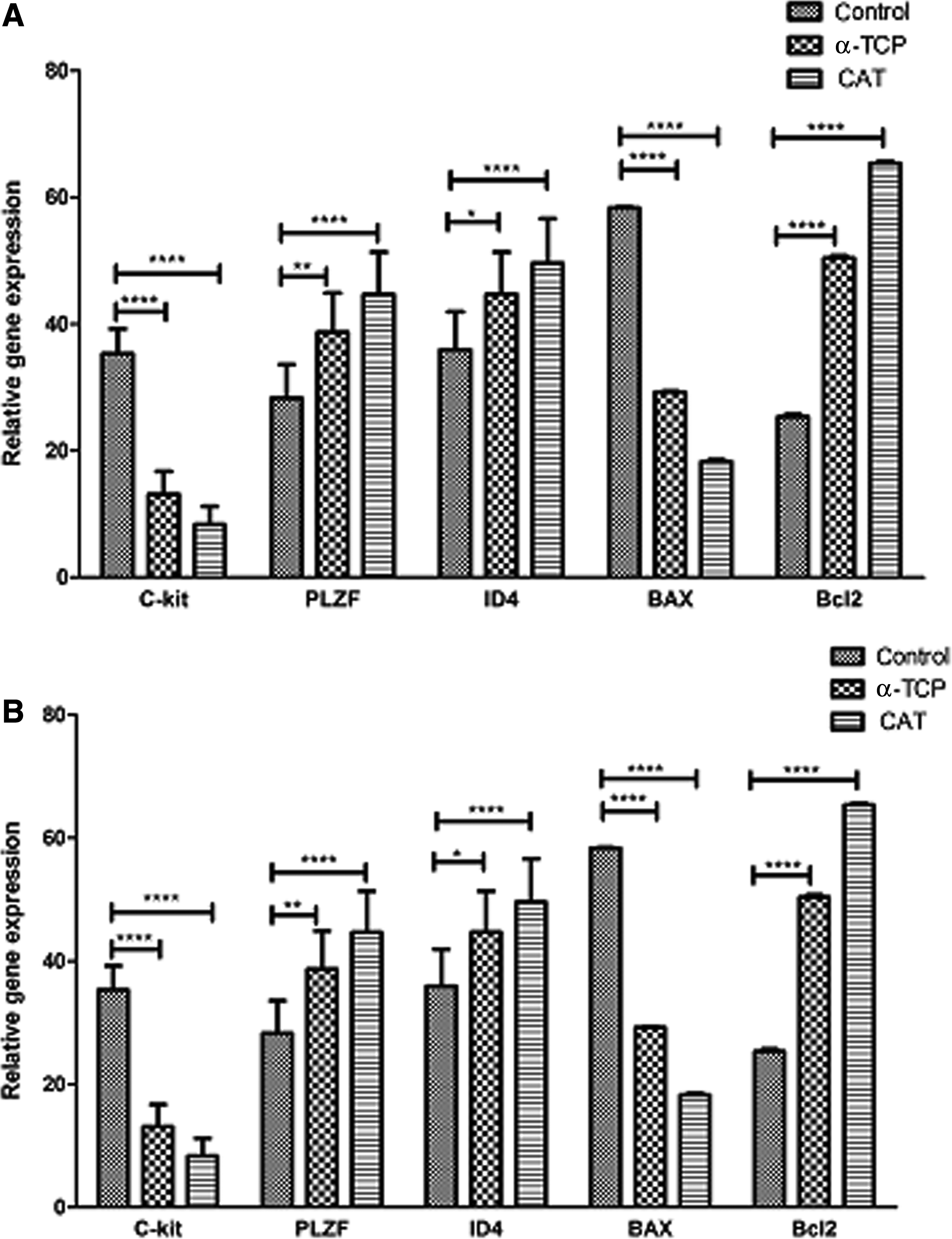
(
Transplantation
To study the survival of SSCs and restoration of spermatogenesis in seminiferous tubules, SSCs were transplanted into the left testis of recipient of busulfan-treated mice. The right testis was considered as the negative control group. After 2 months, testes were evaluated for the occurrence of spermatogenesis (n = 3 in each group). In the recipient testis, the DII-conjugated SSCs were detected at the site of SSCs injection (Fig. 6A), whereas only a few tubules with spermatogenesis were observed at other locations. By counting, about 79% and 75% of the seminiferous tubules had complete spermatogenesis in the CAT and α-TCP groups at the injection sites, respectively, compared with the control group (without antioxidant), which had spermatogenesis in ∼63% of the seminiferous tubules (Fig. 6B–E). The mean percentage of seminiferous tubules with complete spermatogenesis was recorded (Fig. 7).
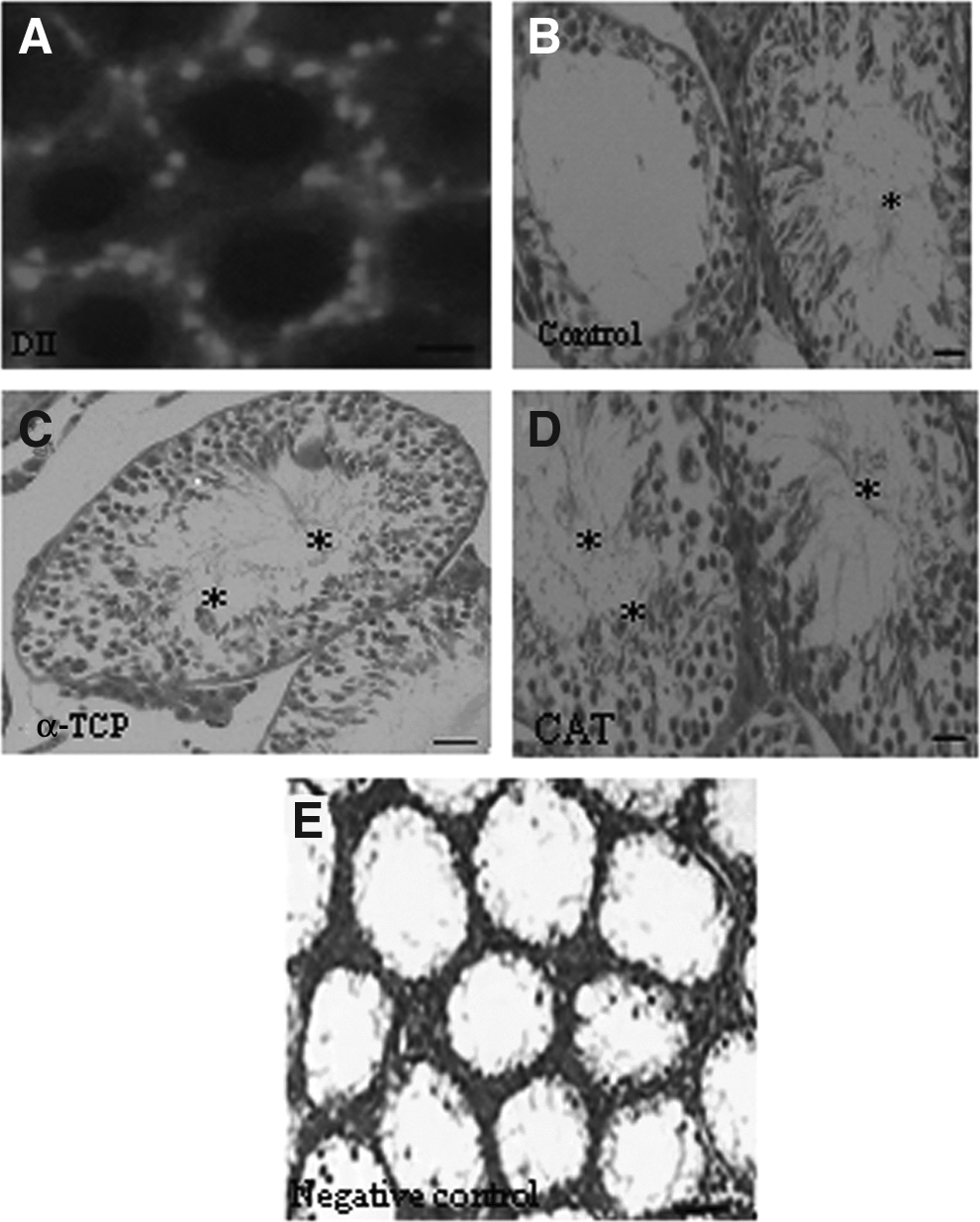
Transplantation of SSCs to recipient mice. Two months after transplantation, recipient testes were analyzed with fluorescent microscope for detecting DII-positive SSCs that were found in the seminiferous tubules (
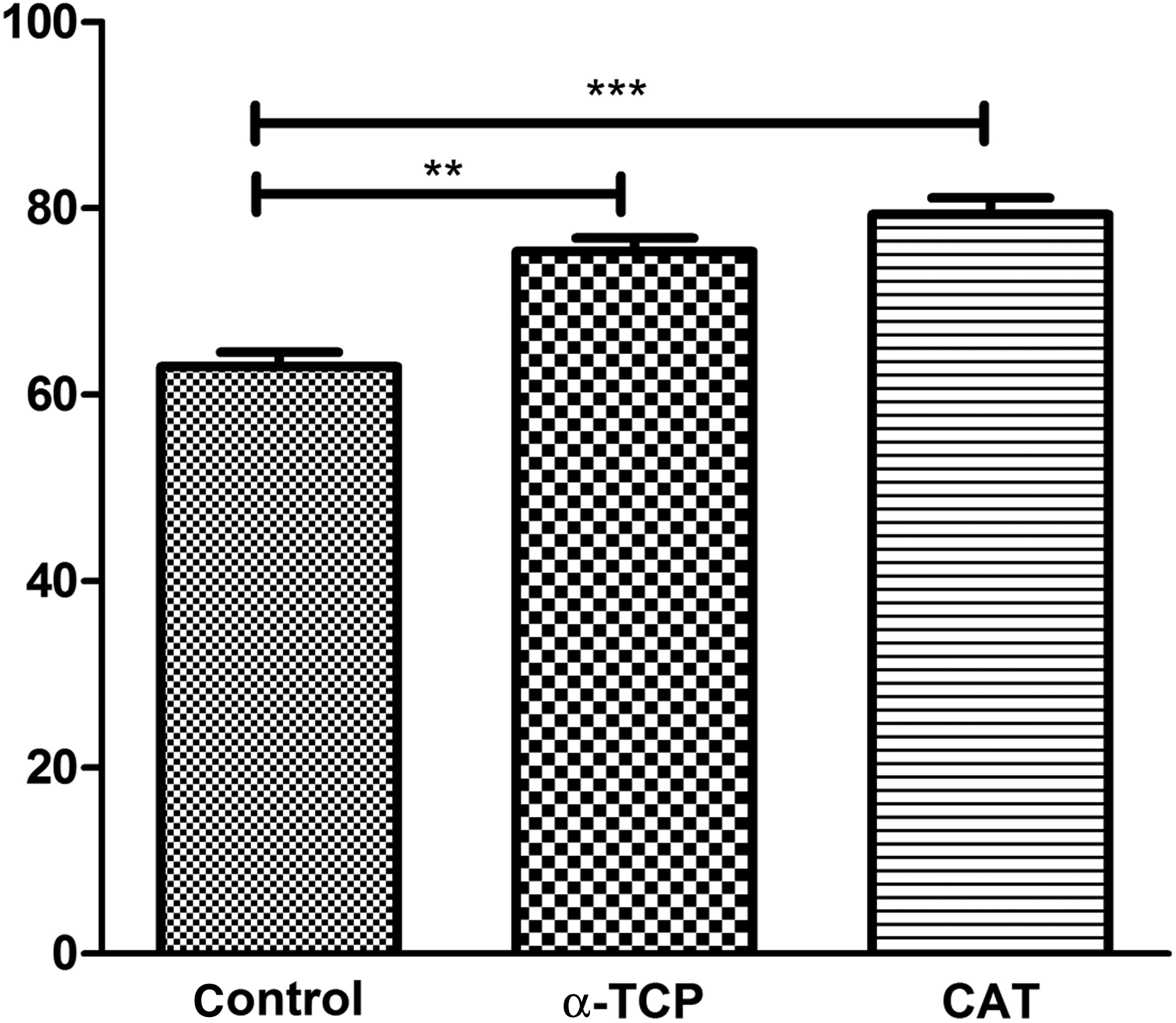
The mean percentage of seminiferous tubules with complete spermatogenesis. Data show means ± SD; (**) p ≤ 0.01, (***) p ≤ 0.001.
Discussion
In this study, we developed a novel cryopreservation protocol for the freezing of mouse SSCs using two different kinds of antioxidants. Our results indicated that cryopreservation of SSCs in the presence of CAT or α-TCP can increase cell viability. After 1 month of culture, SSCs maintained their undifferentiated state and reserved their ability to resume spermatogenesis after transplantation.
For the isolation of SSCs, we used 3- to 6-day-old mice. After enzymatic digestion, the percentage of SSCs was evaluated by a marker of undifferentiated germ cells, Stra8 (Oatley et al., 2008); the isolated cells were then cryopreserved in different conditions. Stra8-positive cells were counted by flow cytometry after isolation and freezing of SSCs in all groups. The higher number of Stra8-positive cells observed in all groups after cryopreservation probably indicates the lethal effects of cooling on Sertoli cells and, thus, a decreasing Sertoli cell population (Koruji et al., 2007). One week after freezing, we analyzed cell viability using the MTT assay. Our results showed that postthaw viability of cryopreserved SSCs was higher when cells were preserved in cryomedium with CAT and α-TCP compared with the control.
Cryopreservation is the best method for long-term preservation of SSCs because it can prevent aging, contamination, and genetic changes in cell lines (Mirzapour et al., 2013). In this study, the method of Izadyar et al. was used for cryopreservation of mice SSCs. They demonstrated that by the addition of cryoprotective agents to cryopreservation media improved the efficiency of cryopreservation of bovine SSCs, and the use of cryoprotective agents improved the recovery of viable cells after thawing (Izadyar et al., 2002). Although cryopreservation is better than culture for long-term preservation of SSCs, the procedure does not come without limitations. One of these limitations is the inability to retrieve successfully adequate numbers of functional SSCs following freezing. One study showed that cryopreservation of bovine stem cells without appropriate cryoprotective agents can lead to cell death by the production of both intra- and extracellular ice (Jahnukainen et al. 2011; Wyns et al. 2010).
The main objective of this study was to develop a more effective cryopreservation protocol for long-term preservation of mice SSCs. To achieve this goal, we evaluated different cryopreservation methods using the antioxidants CAT and α-TCP. Our results showed improvement in efficiency compared with those observed in a previous study in which cell freezing was used to preserve porcine SSCs (Koruji et al., 2007).
After freezing, we cultured postthawed SSCs treated with antioxidants for 1 month. The cells were then evaluated for apoptosis and undifferentiated SSCs markers and the capacity to generate donor colonies of spermatogenesis after transplantation into recipient testes. In this study, the number of colonies and their diameters in CAT and α-TCP cryopreserved groups were better than the control group. This may be attributed to the fact that cryopreservation with antioxidants can probably improve cell enrichment and increase the efficiency of colony formation in isolated SSCs (Koruji et al., 2007; Mirzapour et al., 2013). After 1 month of culture, we analyzed Bax and Bcl2 gene expression, and our data clearly showed that the apoptotic rate was decreased in the presence of antioxidants. Antioxidants have the ability to convert H2O2 to H2O and O2 and eliminate potential ROS toxicity. It seems that by adding antioxidants to the freezing medium, ROS formation can be decreased and the damaging effects of cryopreservation on cells is reduced (Koruji et al., 2007).
Spermatogonial-derived colonies showed SSC marker activity, increased Plzf and ID4 expression, and decreased c-kit expression in both CAT and α-TCP groups. Mirzapour et al. evaluated the effects of cryopreservation on viability, proliferation, and colony formation of human SSCs in vitro culture. They reported that the number and diameter of colonies increased after 3 weeks of culture (Mirzapour et al., 2013). Our results are in agreement with previous studies that reported cryopreservation can preserve purified SSCs, because these cells are relatively resistant to freezing solution compared with other spermatogenic cells (Redden et al., 2009).
It has been suggested that SSCs obtained from freeze–thaw cycles can be used for infertility treatment and transplantation into infertile patients (Kanatsu‐Shinohara et al., 2003). Brinster et al. also demonstrated that freeze–thaw stem cells have a potential to initiate spermatogenesis (Brinster and Zimmermann, 1994). Our findings show that spermatogenesis occurred successfully in recipient mice in all groups; however, spermatogenesis in the CAT and α-TCP groups was better than in the control group. The increase in spermatogenesis in the CAT and α-TCP groups confirms that cryopreservation of SSCs using antioxidants can enhance the possibility of banking SSCs for men who have a malignant disease and are waiting for radiotherapy. Our findings demonstrate that this novel approach of cryopreservation of SSCs with an antioxidant can improve the efficiency of cryopreservation as well as testis cell survival, proliferation in culture, and resumption of spermatogenesis in recipients.
Conclusion
Our results suggest that cryopreservation of SSCs with the protocol reported here can induce SSCs proliferation after 1 month of culture and improve their ability to maintain their undifferentiated state. This new approach in stem cells cryopreservation can be a promising strategy in fertility preservation for patients who suffer from malignancy.
Footnotes
Acknowledgment
We are grateful for the funding support from Tehran University of Medical Sciences, (grant no. 20163). The results described in this paper were part of a student thesis.
Author Disclosure Statement
The authors declare that no conflicting financial interests exist.