
Abstract
Abstract
Primordial germ cells (PGCs) are the undifferentiated progenitors of gametes. Germline competent PGCs can be developed as a cell-based system for genetic modification in chickens, which provides a valuable tool for transgenic technology with both research and industrial applications. This implies manipulation of PGCs, which, in recent years, encouraged a lot of research focused on the study of PGCs and the way of improving their culture. The aryl hydrocarbon receptor (AHR) is a ligand-activated transcription factor that besides mediating toxic responses to environmental contaminants plays pivotal physiological roles in various biological processes. Since a novel compound that acts as an antagonist of this receptor has been reported to promote expansion of hematopoietic stem cells, we conducted the present study with the aim of determining whether addition of an established AHR antagonist to the standard culture medium used nowadays for in vitro chicken PGCs culture improves ex vivo expansion. We have found that addition of α-naphthoflavone in culture medium promotes the amplification of undifferentiated cells and that this effect is exerted by the blockade of AHR action. Our results constitute the first report of the successful use of a readily available AHR antagonist to improve avian PGCs expansion, and they further extend the knowledge of the effects of AHR modulation in undifferentiated cells.
Introduction
P
The manipulation of PGCs is very useful for producing transgenic chickens and also for preserving genetic material in avian species (Nakamura et al., 2010, 2013; Nishijima and Iijima, 2013). A lot of research has focused, therefore, on the study of PGCs and the way of improving their culture, since the major obstacle is to amplify these cells without losing their pluripotent properties. In vitro long-term cultures of germline competent avian PGCs maintained in well-characterized conditions have been achieved (Han, 2009; Han et al., 2002; Naito et al., 2015; Raucci et al., 2015), and the use of PGCs in transgenic experiments for effective production of germline chimera has been established (Han, 2009; Han et al., 2002; Nishijima and Iijima, 2013). Considering the feasibility of current biotechnologies in the chicken, improvements in the maintenance of PGC characteristics for an extended period and the ability to promote their amplification or expansion further contribute toward improving effective systems for germline chimera production and avian genome manipulation.
It has been reported that a purine derivate is able to promote the expansion of human hematopoietic stem cells, and that this effect is exerted by antagonizing the aryl hydrocarbon receptor (AHR) (Boitano et al., 2010). This receptor is a ligand-activated transcription factor that mediates most of the toxic effects of environmental contaminants (Sorg, 2014). As we and others have demonstrated, the AHR exerts pleiotropic responses and diverse effects on reproductive, developmental, nervous, and immune systems (Esser and Rannug, 2015; Hernández-Ochoa et al., 2009; King-Heiden et al., 2012). The expression of this receptor has been described in chicken PGCs, and its involvement in toxicological inhibition of meiosis has been reported (Ge et al., 2012). However, in addition to the toxicological actions of AHR activation, this receptor is also responsive to natural compounds and endogenous physiological signals and plays important roles in the maintenance of homeostatic function, including stem cell cycling, regulation, and quiescence (Gasiewicz et al., 2014; Hao and Whitelaw, 2013).
Given the antecedents outlined earlier, the aim of the present study was to investigate whether AHR antagonism is able to promote the in vitro expansion of avian PGCs, as assessed by determining expression of pluripotency markers in control and treated cell cultures. We demonstrate for the first time that the AHR antagonist α-naphthoflavone promotes further expansion of chicken PGCs maintained in vitro in standard culture conditions and that this positive effect concurred with a strong AHR antagonism that led to complete blockade of AHR transcriptional action.
Materials and Methods
Experimental animals and animal care
Fertilized eggs from a Cobb breeding flock were facilitated by Tres Arroyos Farm (Buenos Aires, Argentina) and incubated at 38.5°C with a relative humidity of 60%. The procedures for animal management and embryo manipulation adhered to the standard operating protocols of our laboratory and were reviewed and approved by the Animal Research Committee of our Institution, which follows the guidelines of the National Institutes of Health, the United States.
Isolation and culture of chicken PGCs
Cobb chicken (Gallus gallus domesticus) embryos at Stage 28 (H&H, 5.5 days of incubation) were collected and rinsed with phosphate-buffered saline (PBS) to remove residual yolk. Gonads were detached with tweezers after a medial section of the abdomen and subsequent dissection under a stereomicroscope. Gonadal cells, which included the PGCs, were isolated and cultured according to the method described by Han et al. (2002). PGCs cultures were maintained in a CO2 incubator at 38°C in Dulbecco's minimal essential medium (DMEM) (Gibco Invitrogen) that was supplemented with 10% (vol/vol) fetal bovine serum (FBS) (Hyclone), 2% (vol/vol) chicken serum (Gibco Invitrogen), 1× antibiotic–antimycotic supplement (Gibco Invitrogen), 10 mM nonessential amino acids (Gibco Invitrogen), 10 mM HEPES (Gibco Invitrogen), 0.55 mM β-mercaptoethanol (Gibco Invitrogen), 2 ng/mL human leukemia inhibitory factor (Sigma-Aldrich Corp.), 0.5 ng/mL human basic fibroblast growth factor (Sigma-Aldrich), 3 ng/mL stem cell factor (Sigma-Aldrich), and 10 ng/mL human insulin-like growth factor-1 (Sigma-Aldrich). From the mixed cell population that was initially seeded, the gonadal stromal cells formed a monolayer whereas the PGCs formed colonies by 7 days of culture. Media were then changed every 5 days, and the isolated colonies of PGCs formed from the mixed cell populations were continuously maintained in a CO2 incubator at 38°C. On day 10, the standard medium described earlier was supplemented with 12 μM α-naphthoflavone (Sigma-Aldrich) in the case of PGCs cultures belonging to the treated group, or an equal volume of vehicle only (ethanol 96%) in the case of control PGCs cultures. Media were changed every 5 days, and incubation was allowed to proceed for 10 additional days.
Alkaline phosphatase staining
Cultured PGCs were fixed in 4% neutral paraformaldehyde for 20 minutes, and alkaline phosphatase (ALP) staining was performed according to the conventional histochemical method with NBT/BCIP (Promega) as the substrate and following the manufacturer's instructions. Incubation was carried out at 37°C for 40 minutes. Quantification of ALP staining was carried out with GeneTools Analysis Software-SynGene Synoptics Ltd. (Cambridge). After cytochemical staining, four different regions were randomly selected in each sample and images were captured with a video camera coupled to a computer. The intensity of staining (influenced by number and area of PGC colonies) was then measured on each image, and the average of the four regions was taken as one replicate. The same procedure was performed for samples coming from both control and treated cultures, in each of the duplicate wells. At least three experiments were made for quantification of differential staining, and results were then statistically analyzed.
Immunofluorescence for stage-specific embryonic antigen 1
Culture media of chicken PGCs were removed, and cells were washed twice with PBS and fixed in P-formaldehyde 4% for 10 minutes. After washing cells with PBS, the cells were permeabilized for 30 minutes in Triton X-100 0.25%, washed afterward with PBS, and blocked for 60 minutes with 10% goat normal serum in PBS–Tween 20 (0.1%). Incubation with the first antibody against stage-specific embryonic antigen 1 (SSEA-1) (mouse IgM; diluted 1:1000; Chemicon®) was done during 2 hours at room temperature. Thereafter, cells were washed with PBS–Tween 20 (0.1%) and incubated with Cy™ three goat anti-mouse IgM (diluted 1:200; Jackson ImmunoResearch, BA, USA) for 1 hour at RT. After washing with PBS, the samples were allowed to dry and then mounted with Mowiol mounting media (Hoechst). Images were then analyzed by confocal microscopy.
Flow cytometry of chicken PGCs
PGCs that had been cultured either in control conditions or in media containing α-naphthoflavone were labeled with anti-SSEA-1 mouse IgM antibody (diluted 1:1000; Chemicon) followed by staining with Cy 2 goat anti-mouse IgM (diluted 1:200; Jackson ImmunoResearch, BA, USA). Incubations were performed in the dark on ice and during 30 minutes. Next, cells were analyzed in an FACS Aria Flow Cytometer (Becton Dickinson) with the analysis gate adjusted to 10,000 single cell events, and data were processed with the WinMDI 2.8 software.
Stable transfection of chicken PGCs
Isolated PGCs that were cultured for 20 days in the described media were transfected with the pIRES2-EGFP plasmid (BD Biosciences Clontech) using 1 μL/cm2 of Lipofectamine 2000® (Invitrogen), 250 ng/cm2 DNA and following the manufacturer's instructions. Twenty-four hours after transfection, culture media were supplemented with 200 μg/mL Geneticin® (G-418; Gibco®) and incubation was allowed to proceed for 5 additional days. After this period of selection, surviving colonies of PGCs that stably expressed the selection marker and the green fluorescent protein were collected and prepared for embryo injection.
Injection of EGFP-transfected PGCs into 2.5-day-old recipient embryos
PGCs cultured in the media described in the preceding sections and stably transfected with the reporter protein EGFP were injected into the bloodstream of 2.5 day recipient embryos. For this purpose, eggs of recipients (a total of ten in each experiment) were incubated at a temperature of 38°C and 60% humidity for 2.5 days until embryos were at stage 17 (H&H). Only eggs with embryos localized centrally were selected for injection, and the remaining embryos (displaced or not appropriately located) were removed due to inconvenient or even impossible access to the blood vessels. Eggs were disinfected with 70% ethanol and drilled in the blunt end. The window generated during this procedure had a diameter of 8 mm. PGCs positive for EGFP were centrifuged and resuspended in PBS, and a total number of 1000 cells in a volume of 10 μL was introduced with a micromanipulator into the bloodstream of embryos through the dorsal aorta. After injection, eggs were sealed with parafilm and incubated for 3 subsequent days at the same standard conditions. All manipulations were done in a biological safety cabinet.
PGCs that remained after microinjection were transferred to a four-well culture plate and cultured to evaluate cell survival rate after the whole procedure. A small fraction was left apart and was subjected to polymerase chain reaction (PCR) to confirm presence of the transgene EGFP.
Detection of EGFP in the gonads of recipient embryos
Gonads were isolated from the 5.5-day-old recipient embryos as described in previous paragraphs, and DNA was isolated and purified from sections of tissue with the Kit illustraTM tissue & cells genomicPrep Mini Spin Kit (GE Healthcare, UK Limited). From each embryo, liver, brain, and muscle were also isolated and DNA purified by following the same procedure. Presence of EGFP transgene was assessed by PCR using specific primers designed with the software Oligo Primers Analysis (Molecular Biology Insights, Inc.). Primer sequences were 5′-GCTGAACTTGT GGCCGTTTAC-3′ (forward primer) and 5′-AGGCCGGTG TGCGTTTG3-′ (reverse primer). These primers amplified a fragment of 623 bp when foreign DNA coming from the injected PGCs EGFP[+] was present in the isolated tissue of recipient embryos. Amplification of target DNA was performed for 41 cycles in the presence of 1.5 mM MgCl2, with each cycle consisting of 15 seconds of denaturation at 94°C, 30 seconds of annealing at 63.6°C, and 30 seconds of elongation at 72°C. The amplification program included an initial step at 94°C for 5 minutes and a final step at 72°C for 3 minutes. Ten microliters of the PCR was electrophoresed in 2% agarose gels with subsequent ethidium bromide staining, and images were acquired with GENi Syngene Synoptics Ltd. Gel Documentation System (Cambridge).
RNA extraction and RT-PCR of PGCs
Levels of CYP1A4 mRNA expression in PGCs were assessed using reverse transcription-polymerase chain reaction (RT-PCR). Cultured PGCs were treated with β-naphthoflavone, α-naphthoflavone, or a combination of both compounds. Control cells received an equal volume of only vehicle (ethanol 96%). After incubation with the different stimuli, cells were lysed directly in the culture dish with TRIzol Reagent (Invitrogen, Molecular Research Center, Inc.) and total RNA was extracted according to the manufacturer's instructions. Complementary DNA was synthesized from total RNA (1 μg RNA in 10 μL of reverse transcription (RT) reaction). A blank without RNA was included in each set of RT reactions. A control of RNA that was not subjected to RT was also included in subsequent PCRs. One microliter aliquots of the RT reaction was used to amplify CYP1A4 and 18S fragments in a multiplex reaction. The primer sequences and PCR protocol and parameters used to amplify the hydroxylase target cDNA were those described by Bussmann et al. (2013). We have previously demonstrated that minor changes in CYP1A mRNA levels are certainly detected with this technique (Bussmann and Barañao, 2008; Bussmann et al., 2006, 2013), and we have shown and established that AHR activation and blockade through antagonist treatment can be definitely verified with this methodological approach and with these techniques in chicken cells (Bussmann et al., 2013).
Statistical analysis
Treatments were applied to at least duplicate wells in each of three separate experiments, unless otherwise indicated. In those experiments where a representative qualitative result is shown, experiments had been carried out at least three times, obtaining similar results. RNA expression experiments: Results are expressed as the mean ± SEM of the independent experiments. Statistical comparisons of the results were made using one-way ANOVA and Tukey–Kramer's test for multiple comparisons after logarithmic transformation of data when necessary (Sokal and Rohlf, 1995). ALP Staining experiments and Flow Cytometry experiments: Results are expressed as the mean ± SEM of the independent experiments. A paired t-Student test was used to compare the values obtained in control and flavone-treated cells (positive staining for ALP activity or expression of SSEA-1) after logarithmic transformation of data when necessary (Sokal and Rohlf, 1995).
Results
Alpha-naphthoflavone increases ALP-positive cells in cultured PGCs
ALP is a universal pluripotent marker for all types of pluripotent stem cells, including embryonic stem cells, embryonic germ cells, and induced pluripotent stem cells. The pluripotent status of stem cells can be characterized by a high level of ALP expression. Therefore, the effect of the AHR ligand α-naphthoflavone on the amplification of cells with ALP activity was determined in our isolated PGCs cultures that had been obtained and maintained as described and validated by Han et al. (2002). As can be seen in Figure 1, left panel, PGC colonies maintained in the standard “embrionic germ” medium showed a strong positive mark for ALP. Compared with these control groups, PGCs after α-naphthoflavone treatment for 10 days showed significant enhanced proliferation, as evidenced by an increase in ALP-positive cells (Fig. 1, right panel). The images shown in Figure 1 are representative pictures of one representative experiment. Densitometric analysis performed with quantification purposes revealed that α-naphthoflavone-treated cultures contained in average ∼40% more ALP-positive cells than control cultures (Fig. 1, lower panel).
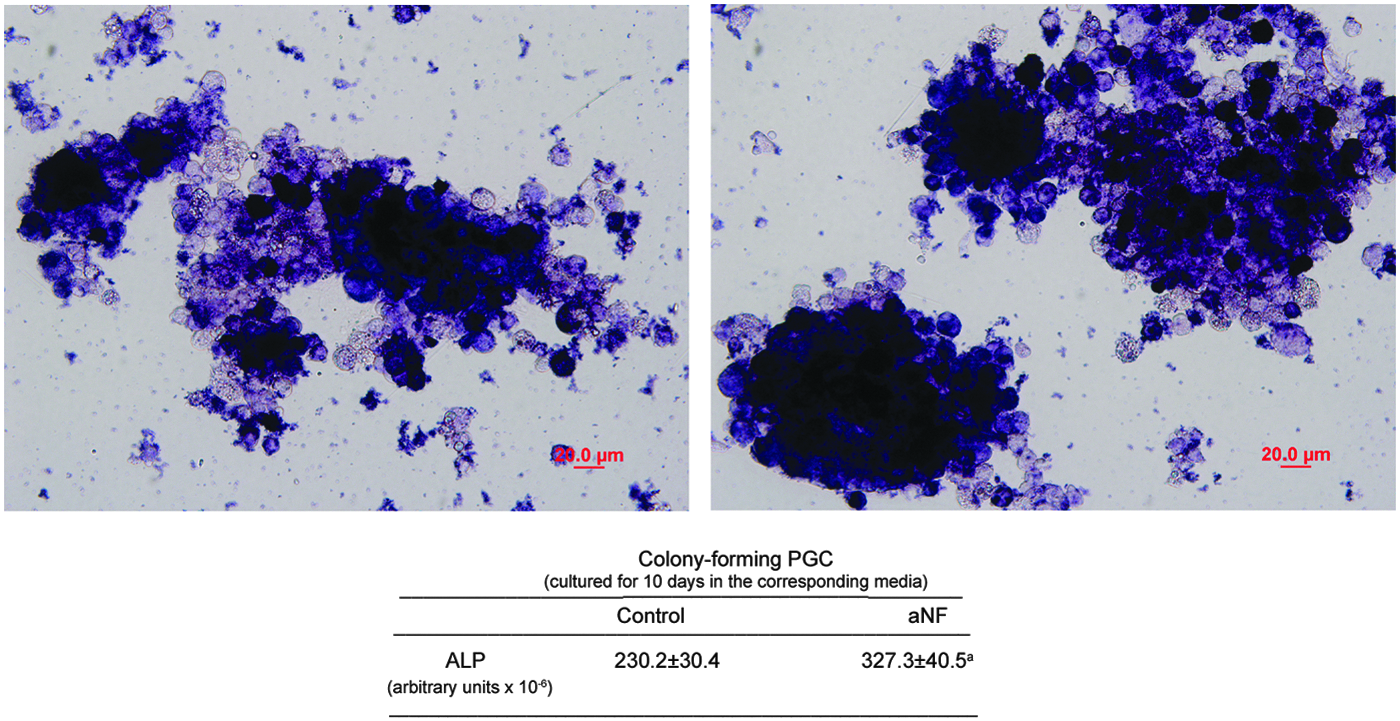
Alpha-naphthoflavone increases the number of cells with ALP activity in cultured PGCs. PGCs were cultured as described in the Materials and Methods section and maintained in control medium (left panel) or in medium containing α-naphthoflavone (aNF, 12 μM, right panel) for an additional period of 10 days. Alkaline phosphatase staining was performed as described in the Materials and Methods section to identify undifferentiated primordial cells. Photographs shown are representative images of one of the three independent experiments performed. Lower panel: Intensity of staining, expressed in arbitrary units as the mean ± SEM of three independent experiments, of cultures maintained in control medium or in medium containing α-naphthoflavone (aNF). aSignificant difference between the staining intensity of control and treated cultures, p < 0.05. PGCs, primordial germ cells. Color images available online at www.liebertpub.com/cell
Alpha-naphthoflavone induces expansion of SSEA-1-positive cells in cultured PGCs
To further evaluate the expansion of undifferentiated primordial cells in the presence of α-naphthoflavone, the expression of another marker for PGCs, namely the SSEA-1, was assessed in control and treated cultures. As shown in Figure 2, treatment of cultured PGCs with the flavone during 10 days resulted in a rise of cells that were positive for this marker, in comparison to control cultures in which the flavone had not been added.
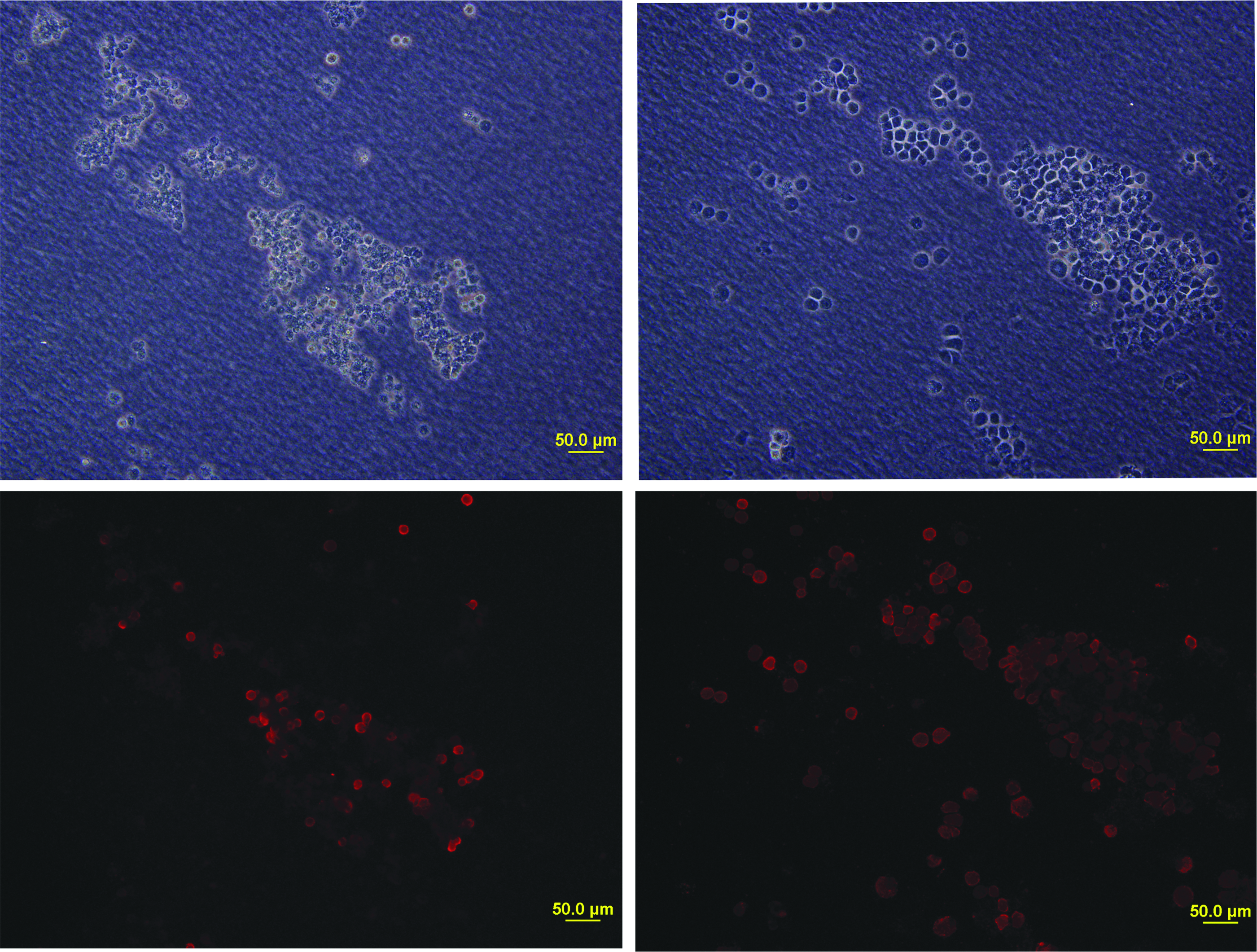
Alpha-naphthoflavone induces SSEA-1+ cell expansion in cultured PGCs. PGCs were cultured as described in the Materials and Methods section and maintained in control medium (left panels) or in medium containing α-naphthoflavone (aNF, 12 μM, right panels) for an additional period of 10 days. Immunofluorescence labeling for SSEA-1 was performed as described in the Materials and Methods section to identify undifferentiated primordial cells (lower panels). Phase-contrast images were performed for clear cell images (upper panels). SSEA-1, stage-specific embryonic antigen 1. Color images available online at www.liebertpub.com/cell
Alpha-naphthoflavone increases the number of cells positive for SSEA-1 in cultured PGCs
With the purpose of accurately quantifying the degree of expansion undergone by undifferentiated primordial cells when α-naphthoflavone is added to cultured PGCs, SSEA-1-labeled cells were subjected to FACS (Fig. 3A, B). Flow cytometry confirmed that a significant increase in cells positive for this marker is observed in cultures treated with the flavone: a mean of 65.0% of cells positive for SSEA-1 in control cultures versus a mean of 87.7% in treated ones (p ≤ 0.05, Fig. 3C).
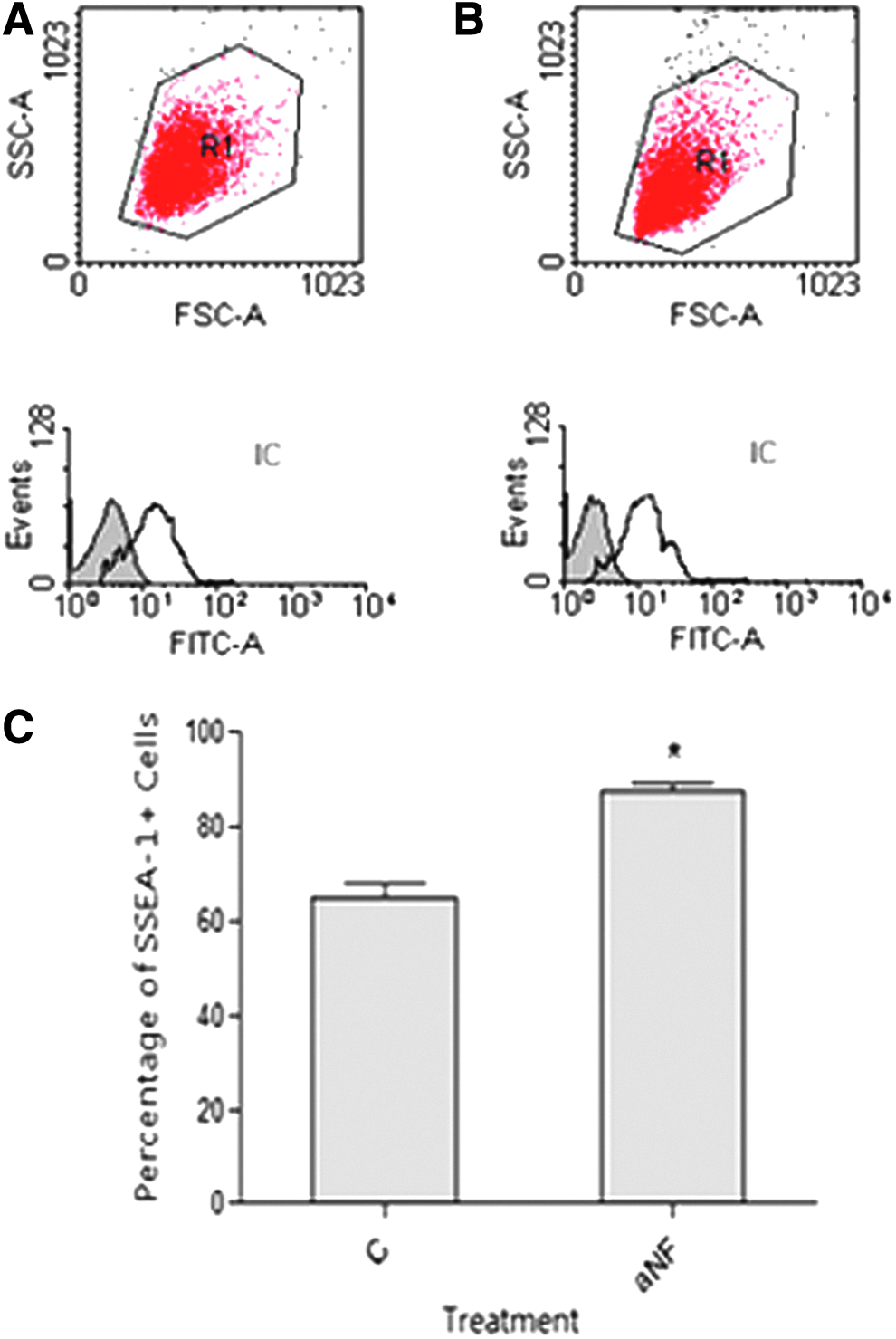
Alpha-naphthoflavone increases the number of SSEA-1+ cells in cultured PGCs. PGCs were cultured as described in the Materials and Methods section and maintained
PGCs cultured in media containing α-naphthoflavone retain their ability to migrate and colonize the genital ridge of recipient embryos
To further characterize the PGCs cultures, we corroborated that the undifferentiated primordial cells maintained in our culture conditions were capable of migration and colonization of the gonadal ridges of recipient embryos. The migratory and colonization ability of PGCs was assessed by injecting in recipient embryos at 2.5 days of incubation cells that had been previously transfected with the reporter green fluorescent protein EGFP. Tracing of the original PGCs was made by detection of the foreign EGFP DNA in the different tissues of the recipient embryo after three additional days of incubation. As can be seen in Figure 4A, foreign DNA specific of EGFP could be detected exclusively in the gonadal tissue of the recipient tissues, and not in other tissues such as muscle, liver, or brain. This evaluation was made in three different independent experiments, each consisting of the injection of 7–10 embryos (eggs that did not meet the inclusion criteria for microinjection were discarded, which explains that in experiments B and C less embryos were included). As can be seen in the table in Figure 4B, microinjection/migration/colonization of the gonadal ridges of recipient embryos by PGCs is made with certain efficiency, specifically 60% in our experiments. It is to be noted, however, that this efficiency depends largely on the efficacy and accuracy of injection, mainly due to the difficult maneuver involved. Besides, and most importantly, the efficiency equals the one obtained when injecting PGCs that had been cultured in standard conditions lacking α-naphthoflavone and both are comparable to those reported by some authors (Jeong et al., 2002).
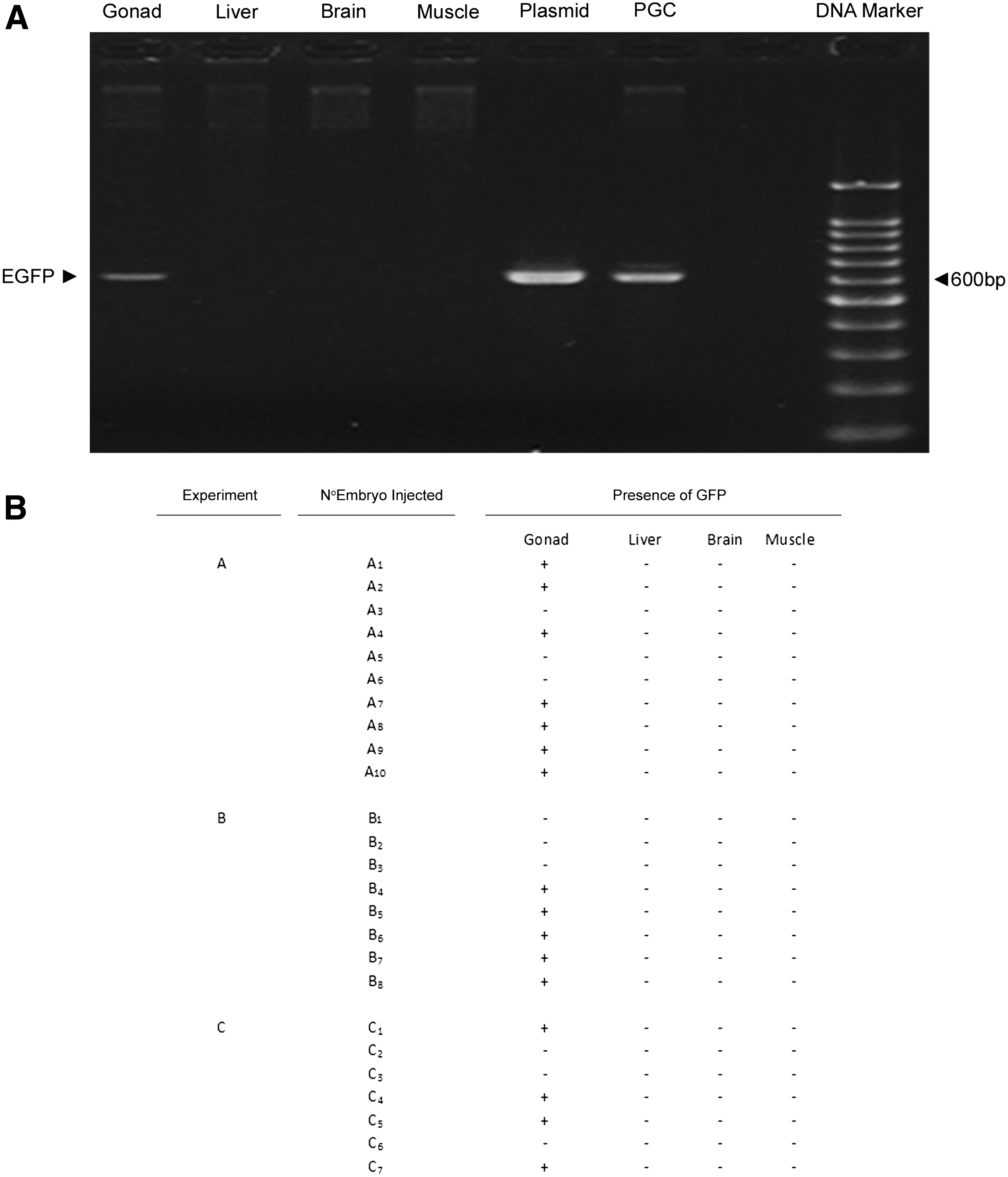
PGCs cultured in media containing α-naphthoflavone retain their migration and colonization ability. PGCs cultured in α-naphthoflavone-containing media and transfected with the green fluorescent protein EGFP were injected in recipient embryos at 2.5 days of incubation as described in the Materials and Methods section. After 3 additional days of incubation, gonadal tissue, liver, brain, and muscle were isolated and DNA was purified as described in the Materials and Methods section. Samples were subjected to PCR to assess the presence of foreign DNA specific for EGFP. Approximately 10 embryos were injected in each experiment, and a total of three independent experiments were carried out.
Alpha-naphthoflavone blocks AHR activation in cultured PGCs
With the aim of determining whether α-naphthoflavone is able to block AHR function and act as an antagonist of this receptor in our system of cultured chicken PGCs, we evaluated the transcriptional activity of this protein. In chickens, Cyp1a4 gene transcription is considered a hallmark of Ah response in most cells, constituting a model for the study of the AHR as transcription factor (Gannon et al., 2000; Mahajan and Rifkind, 1999). Therefore, we assessed the transcript levels of this gene in control conditions and when α-naphthoflavone and/or the classic AHR agonist β-naphthoflavone was added to culture media. As can be seen in Figure 5, α-naphthoflavone is able to reduce the basal endogenous levels of Cyp1a4 transcripts and to completely antagonize the stimulatory effect of β-naphthoflavone on the expression levels of this gene.
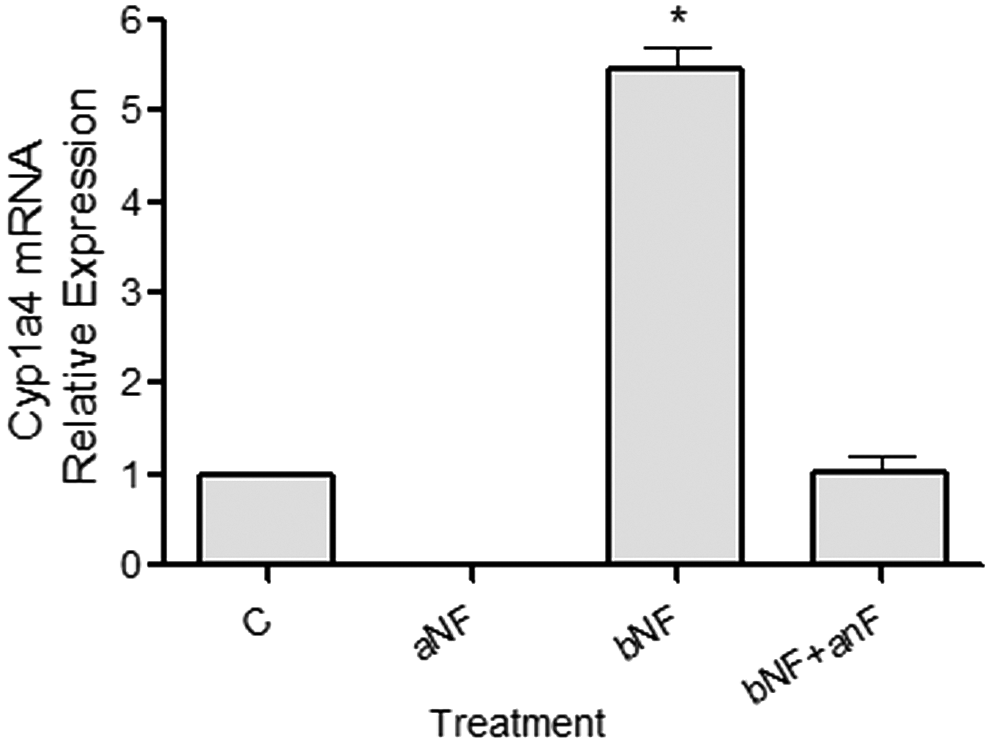
Alpha-naphthoflavone blocks AHR activation. PGCs were cultured as described in the Materials and Methods section for 10 days in control medium (C), in the presence of α-naphthoflavone (aNF, 12 μM), β-naphthoflavone (bNF, 6 μM), or a combination of both AHR ligands. Total RNA extraction and reverse transcription-polymerase chain reaction (RT-PCR) for CYP1A4 mRNA were performed as described in the Materials and Methods section. The amount of each mRNA was normalized to the 18S ribosomal signal for each sample, and values (relative to control cells) were plotted as the mean ± SEM of three independent experiments, depicting the normalized gene expression of control and treated cells. Asterisk denotes significant differences (*p < 0.001). AHR, aryl hydrocarbon receptor; RT-PCR, .
Discussion
The feasibility of chicken PGCs manipulation, including in vitro culture and expansion of these cells, allows transgenic chicken generation and preservation of genetic material in avian species. Therefore, a lot of research has been conducted either to study PGCs and improve their culture (Han, 2009; Nakamura et al., 2013) or to develop new approaches and offer alternative protocols for transgenesis (Yu et al., 2014). Findings of a positive effect of AHR antagonists on the maintenance of hematopoietic stem cells in culture (Boitano et al., 2010; Carlin et al., 2013; Gasiewicz et al., 2014) led us to investigate the action of α-naphthoflavone on the in vitro expansion of chicken PGCs. To this end, we evaluated the effect of this flavone on the pluripotent status of our cultures, as assessed by changes in levels of ALP and SSEA-1 expression. In this study, we have found that addition of this flavone in culture medium promotes the expansion of undifferentiated cells and that this effect is exerted by antagonizing AHR action. To the best of our knowledge, this constitutes the first report showing a beneficial effect of an AHR antagonist on PGCs culture.
The results found in this study are in accordance with those reported for other undifferentiated cells such as hematopoietic stem cells (Boitano et al., 2010; Carlin et al., 2013; Gasiewicz et al., 2014) and likewise suggest that the AHR is partially active in PGCs, where it functions to drive cells toward differentiation. This is evident in our experiments where AHR transcriptional activity is assessed in culture PGCs, since addition of α-naphthoflavone inhibits not only the agonist-induced transcript levels but also the basal ones. As none of the factors present in the culture medium are known AHR agonists, the observed basal activation of the receptor in our system might indicate the presence of an endogenous ligand for this receptor in PGCs. There is enough evidence to postulate that the AHR has a normal function in the regulation of hematopoietic and other stem/progenitor cell populations (Singh et al., 2009), and our data add support to this notion.
Although the mechanistic explanations that account for the effect of α-naphthoflavone described here are beyond the aim of the present study, future research regarding this question is warranted. It has been postulated that the AHR could drive pluripotent cells toward differentiation through its ability to downregulate the stem cell transcription factor Oct4 (Bunaciu and Yen, 2011), which would be a possible mechanism underlying the induction of proliferation and the maintenance of the undifferentiated state of chicken PGCs that we observed after AHR blockade. Besides, it has been demonstrated in an in vitro model of induced gametogenesis that toxicological activation of AHR through PAH exposure promotes the apoptosis of chicken PGCs (Ge et al., 2012). This prompts us to speculate that apoptosis would be an alternative or additional mechanism underlying this possible physiological role of the AHR, normally acting as a negative regulator to restrain or limit an excessive or unnecessary proportion of pluripotent cells.
Besides the quantifiable effect of α-naphthoflavone on the number of cells expressing pluripotency markers, we could observe that cultures treated with the AHR antagonist showed improved quality and could be maintained for longer periods, as determined by means of morphological assessment (De Felici and McLaren, 1982). Although the potential mechanism underlying this effect of the flavone remains to be elucidated, it is consistent with the notion that AHR repression leads to loss of quiescence and enhanced self-renewal in undifferentiated stem-like cells (Gasiewicz et al., 2014). In this regard, it has been demonstrated that polychlorinated biphenyls, a group of compounds that function as AHR agonists, decrease PGCs number dose dependently and exert other damaging effects on chicken PGCs (Fang et al., 2002; Zhang et al., 2005).
In the present study, we isolated and cultured chicken PGCs according to validated methods previously described and we obtained comparable efficiencies in culturing cells that maintain stem cell characteristics (Han et al., 2002). However, the medium containing α-naphthoflavone displayed a stimulatory action on PGCs proliferation/survival that caused a rise in the proportion of undifferentiated cells in culture. These cells, after appropriate labeling and harvesting, were able to colonize the genital ridges of recipient embryos after injection into the blood vessels at 2.5 days of incubation, which further characterizes our cultures, confirming their primordial germ status and verifying that the presence of α-naphthoflavone does not affect PGC migratory ability. Our findings are of special interest, because they prove that addition of α-naphthoflavone, a readily available and an inexpensive compound, improves chicken PGCs ex vivo expansion, which can be used to improve any existing protocol used to induce proliferation and maintain avian PGCs. This takes on special relevance for rare or endangered species, in which case the source of PGCs is limited. Furthermore, insights in chicken PGCs in vitro maintenance serve to identify putative factors that are useful for improvements in maintaining self-renewal and pluripotency in mammalian/human PGCs.
Footnotes
Acknowledgments
The authors gratefully acknowledge the generous contribution of Dr. M. Plano from Tres Arroyos Farm (Buenos Aires, Argentina), who kindly facilitated the fertilized eggs. This work was supported by the ANPCYT and CONICET through grants PICT 2008-1175 and PIP 112-200801-03201, respectively.
Author Disclosure Statement
The authors declare that no conflicting financial interests exist.