
Abstract
Abstract
The myocyte enhancer factor-2 (MEF2) is a member of the MADS-box family. It controls the expression of genes that are critical for biological processes such as proliferation, cell death, and differentiation. Some studies have shown that MEF2 expression is enhanced in osteogenic progenitor cells established from bone marrow stromal cells with other types of mesenchymal progenitor cells. However, the effect of MEF2 on dental pulp stem cells (DPSCs) is unclear. In this study, we investigate the effect of MEF2 on regulating osteogenic differentiation and proliferation of DPSCs. We find that MEF2 is stably expressed in DPSCs, and the expression is increased time-dependently along with cell osteogenic differentiation. MEF2 expression also increases the alkaline phosphatase (ALP), runt-related transcription factor 2 (Runx2) activity, and enhances mineralization in DPSCs. SB202190, inhibitor of p38, blocks the p38/MEF2 pathway and osteogenic differentiation. In addition, MEF2 overexpression inhibits DPSC proliferation. In summary, our data indicate that MEF2 not only regulates DPSCs as an inhibitor of cell proliferation but is also a promoter of osteogenic differentiation through the p38/MEF2 signaling pathway.
Introduction
D
Comparison of their multipotency with bone marrow stem cells (MSCs) has demonstrated that proliferation, availability, and cell number of DPSCs were greater than for bone marrow MSCs (Alge et al., 2010; Huang et al., 2009). Moreover, DPSCs can easily be isolated from the human pulp tissue and do not cause ethical and technical problems (Rodríguez-Lozano et al., 2012). Thus, determination of the molecular responsible signal of DPSC differentiation in the process may provide a powerful tool for clinicians on the appropriate application of regenerative therapy for regeneration of these cells.
The four myocyte enhancer factor-2 (MEF2) proteins (MEF2A-D) belong to the minichromosome maintenance 1-agamous-deficiens-serum response factor (MADS)-box family of transcription factors and they have important roles in proliferation and differentiation in organisms as diverse as plants, fungi, and metazoans (Naya and Olson, 1999; Theißen et al., 1996; Yu et al., 1992). MEF2, as an important transcription factor, has diversity of functions in a wide range of cells and has been implicated in many diseases. It is highly expressed in muscle cells, where it has been shown to be an important regulator in the process of gene expression during development of skeletal, cardiac, and smooth muscle (Naya and Olson, 1999). MEF2 protein has also been correlated with the vertebrate developmental stages of neuronal maintenance, survival, proliferation, and differentiation (Heidenreich and Linseman, 2004). The MEF2C showed enhanced expression in a cell line of murine osteogenic progenitor cells, Kusa, was established from femoral bone marrow stromal cells (Kawashima et al., 2005).
Moreover, mitogen-activated protein kinase (MAPK) signaling can enhance the MEF2 transcriptional activity of various cell types. In the cardiac muscle cells, activating the MAPK family member p38 enhances the MEF2C transactivation activity (Han et al., 1997). In skeletal muscle cells and nerve cells, the MAPK/p38 pathway, to regulate MEF2 transcription activity, stimulates the differentiation of skeletal muscle to prevent nerve cell apoptosis (Lu et al., 2000; Mao et al., 1999; Puri et al., 2000; Zetser et al., 1999). However, it seems that the P38/MEF2 pathway has not been reported in the field of osteogenesis. Recently, further roles and relationships of MEF2 have been found, such as its action as an apoptotic factor in neurogenesis and stem cell regulation (Okamoto et al., 2000, 2002). However, the role of MEF2 in regulating DPSCs remains unclear.
In this work, we have examined the effect of MEF2 that regulates proliferation of DPSCs and the progression of osteoblast differentiation and the p38/MEF2 pathway. We find that MEF2 overexpression can promote osteogenic differentiation of DPSCs blocked by p38 inhibitor, SB202190. Our finding may provide a foundation for enhancing osteoblast differentiation in DPSCs promoting bone regeneration.
Materials and Methods
Induction of osteogenic differentiation
DPSCs were plated at a density of 2 × 104 cells/cm2 and cultured in the growth medium to undergo differentiation stimulation. The differentiation medium was supplemented with 10 nM dexamethasone (Sigma), 10 mM β-glycerophosphate (Sigma), and 50 μg/mL ascorbic acid (Sigma). DPSCs were differentiated for 3, 5, 7, and 14 days. The degree of extracellular matrix calcification was estimated using Alizarin red S (Sigma) and alkaline phosphatase (ALP; SenBeiJia) staining. This time point was considered as day 0. Cells were maintained with the addition of fresh osteogenic induction medium every 3 days.
Cell culture
Normal human impacted third molars were collected from patients 13 to 23 years of age (n = 9) after giving the informed consents, which were approved by the Ethics Committee of the Affiliated Hospital of Nantong University. All subjects were free of carious lesions and oral infection. We isolated DPSCs by cleaning the tooth surface, cutting around the cementoenamel junction using sterilized dental fissure burs, and then opening to reveal the pulp chamber. The pulp was digested in a solution of 3 mg/mL collagenase type I for 1 hour at 37°C. Single-cell suspensions were obtained by passing the digested tissues through a 70-μm cell strainer (BD Falcon). Cell suspensions of dental pulp were seeded into 25-cm2 culture dishes and cultured in Dulbecco's modified Eagle medium supplemented with 10% fetal bovine serum (FBS), 100 U/mL penicillin, and 100 μg/mL streptomycin at 37°C in 5% CO2. The medium was changed every 3 days. Cells were passaged at the ratio of 1:3 when they reached 85%–90% confluence. The cell populations were characterized by positive staining with anti-CD34, STRO-1, and c-kit and the absence of CD45 (date not show). Cells at passage 3 (P3) were used in the subsequent experiments.
Assay of ALP activity and mineralization
DPSCs were fixed with 4% PFA for 1 hour and washed with phosphate-buffered saline (PBS). Two percent Alizarin red S solution was applied and DPSCs were placed in an incubator at 37°C for 2 hours. Mineralization was quantified by extracting the Alizarin red S stain with 100 mM cetylpyridinium chloride (Sigma-Aldrich) at room temperature for 2 hours. Absorbance of the extracted Alizarin red S stain was measured at 570 nm. DPSCs were subjected to ALP staining using the ALP assay kit according to the manufacturer's instructions. ALP and Alizarin red S activities were determined by a colorimetric assay.
Reverse transcription and real-time PCR analysis
Total cellular RNA was extracted from cells and reverse transcribed using conventional protocols. PCR amplification was performed using the following primer sets: glyceraldehyde-3-phosphate dehydrogenase (GAPDH) 5′-TCCATGACAACTTTGGTATCG-3′, 5′-TGTAGCCAAATTCGTTGTCA-3′; ALP 5′-TGAAATATGCCCTGGAGC-3′, 5′-TCACGTTGTTCCTGTTTAG-3′; MEF2 5′-GACGCGTAAAACGGGGACTATGGGGAG-3′, 5′-CGGGATCCTTGAACCTGCAACATGACACTATC-3′; and runt-related transcription factor 2 (Runx2) 5′-CTCACTACCACACCTACCTG-3′, 5′-TCAATATGGTCGCCAAACAGATTC-3′. All the primer sequences were determined using established GenBank sequence. The primers were used to amplify the duplicate PCR reactions. Each sample was analyzed in triplicate and GAPDH was used as a control.
Western blot analysis
DPSCs were cultured under appropriate conditions, washed with ice-cold PBS, and cells were lysed in lysis buffer containing 50 mM Tris-HCl, 150 mM NaCl, 2% sodium dodecyl sulfate (SDS), and a protease inhibitor mixture. After centrifugation at 12,000 rpm for 12 minutes, protein concentrations were determined using the Bradford assay (Bio-Rad). The resulting supernatant (50 μg of protein) was subjected to SDS-polyacrylamide gel electrophoresis. The separated proteins were transferred onto PVDF membranes at 350 mA for 2.5 hours in a blotting apparatus (Bio-Rad). Membranes were blocked with 5% nonfat milk and the membranes were immunoblotted with various antibodies (1:1000) at 4°C overnight and subsequently with anti-rabbit horseradish peroxidase-conjugated secondary antibodies (1:10,000; sigma) for 2 hours at room temperature. Concomitantly, GAPDH was run as a reference protein. The following primary antibodies were used: GAPDH (anti-rabbit; Santa Cruz), MEF2 (anti-rabbit; Santa Cruz), ALP (anti-rabbit; Santa Cruz), p38 (anti-rabbit; Santa Cruz), p-p38 (anti-rabbit; Santa Cruz), and Runx2 (anti-rabbit; Santa Cruz).
Immunofluorescence staining
MEF2 expression in DPSCs was examined by immunofluorescence staining. Cells were seeded in 24-well plates at a density of 2 × 104 cells. After incubation for 24 hours, DPSCs were fixed with 4% PFA for 1 hour, washed using PBS containing 0.1% Triton X-100 (PBST) for 15 minutes at room temperature, and blocked for 30 minutes in PBST supplemented with 10% FBS. Anti-MEF2 antibody (1:400) was added and incubated overnight at 4°C. Cells were washed and incubated in second antibody (1:200) for 2 hours at 37°C. Nuclei were stained with 4′,6′-diamidino-2-phenylindole dihydrochloride (DAPI, 1:800; Santa Cruz). The cells were examined using a Leica fluorescence microscope.
MTT colorimetric assay
DPSCs were plated at a cell density of 3 × 103 cell/well in 96-well plates. After incubation for 24 or 48 hours, the proliferation rate of the cells was assessed by MTT (3-dimethylthiazol-2, 5-diphenyltetrazolium bromide; Sigma) test. The culture medium was added with 50 mL of MTT (50 mg/mL; Sigma) to each well and the cells were incubated for 4 hours at 37°C under 5% CO2. The precipitate was extracted with DMSO and the plates were read on a microplate reader at a wavelength of 540 nm.
MEF2 RNA silencing
The siRNA specific for MEF2 was commercially synthesized (Genpharm). For controls, scrambled RNA oligonucleotides were used. For each well, 100 nM siRNA was transfected into the cells using Lipofectamine 2000 (Invitrogen) according to the manufacturer's instructions. Overnight, transfected cells were used for subsequent experiments.
Plasmid constructs and plasmid transfection
The full-length MEF2 (Genbank Accession No. NM 001273913.2) was extracted from the human cDNA library and connected to P-CMV-Flag. The primers used for MEF2 were as follows: 5′-GACGCGTAAAACGGGGACTATGGGGAG-3′ (sense), 5′-CGGGATCCTTGAACCTGCAACATGACACTATC-3′ (anti-sense). Transfection of P-CMV-Flag and P-CMV-Flag-MEF2 in DPSCs was carried out with Lipofectamine 2000 (Invitrogen) according to the manufacturer's instructions.
Statistical analyses
Data are shown in averages ± standard deviations. Statistical analyses were performed with one-way repeated-measures analysis of variance and Bonferroni post hoc test by using KaleidaGraph 4 (HULINKS). Values of p < 0.05 were regarded as significant.
Results
MEF2 is expressed in DPSCs and expression is increased time-dependently in osteogenic differentiation
To understand the role of MEF2 in osteoblast, we examined its expression in osteogenic induction medium. DPSCs were cultured under conditions that promote differentiation for 3, 5, 7, and 14 days. The protein concentrations of MEF2 were positively expressed in the DPSCs (Fig. 1A, B). MEF2 mRNA was also increased time-dependently (Fig. 1C) (*p < 0.05). Since the expression of MEF2 was increased in the process of osteogenic differentiation of DPSCs, we suspected if the overexpression of MEF2 in cells can promote the process of differentiation. We continued to detect the expression of osteogenic differentiation-related genes, ALP and Runx2. We saw that the expression of ALP and Runx2 increased gradually with the increase in days of differentiation (Fig. 1D, E) (*p < 0.05). Then, we transfected MEF2 into DPSCs on fifth day of differentiation; we observed that the levels of ALP and Runx2 reached normal levels of differentiation on the seventh day (Fig. 1F, G) (*p < 0.05).This suggests us that MEF2 can accelerate the process of osteogenic differentiation of DPSCs.
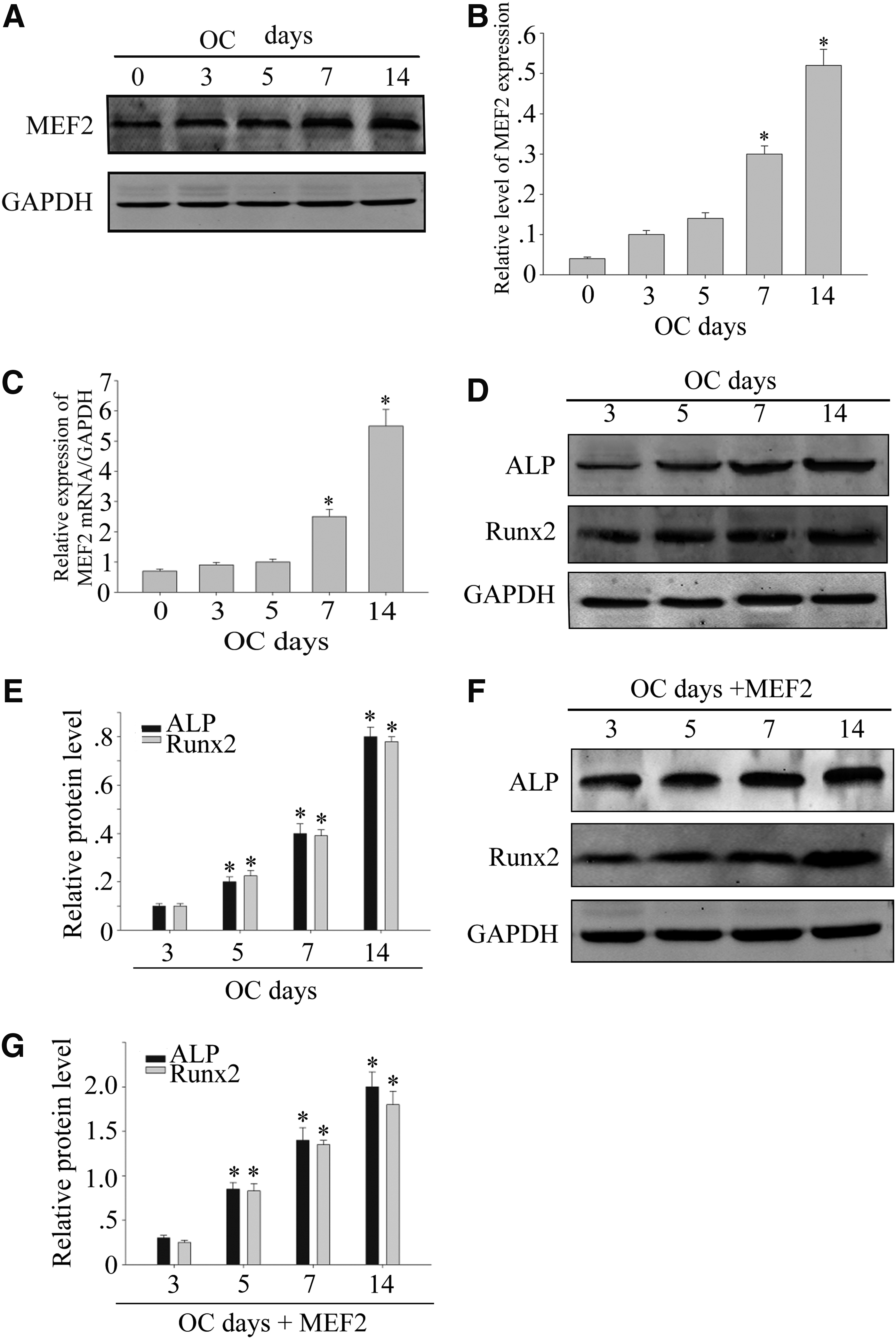
MEF2 expression in osteoblast differentiation of DPSCs.
MEF2 overexpression in DPSCs by stable transduction and enhanced osteogenesis
To determine the effect of MEF2 on DPSC proliferation and differentiation, we transfected a full-length MEF2 cDNA into DPSCs. MEF2 expression in transfected DPSCs was detected by Western blotting and RT-PCR. MEF2 protein level was detected in the MEF2-transfected cells, and the level was low in the untreated and Flag-transfected cells (Fig. 2A) (*p < 0.05). MEF2 mRNA expression was significantly increased in MEF2-transfected DPSCs when compared with untreated and Flag-transfected cells (Fig. 2B) (*p < 0.05). Furthermore, the immunocytochemistry of MEF2 was detected. MEF2 had a higher intensity in the cellular nucleus in MEF2-transfected cells (Fig. 2C). Then, after 14 days of incubation, Alizarin red S staining for mineralized matrix results showed that MEF2 significantly increased mineralized nodules (Fig. 2D) (*p < 0.05). After 7 days of incubation, ALP staining showed that after transfected MEF2, ALP activity was also increased (Fig. 2E) (*p < 0.05). Similarly, RT-PCR analysis revealed that ALP and Runx2 mRNA levels were high in MEF2-transfected cells (Fig. 2F) (*p < 0.05). These findings support that MEF2 overexpression promotes osteoblast differentiation.
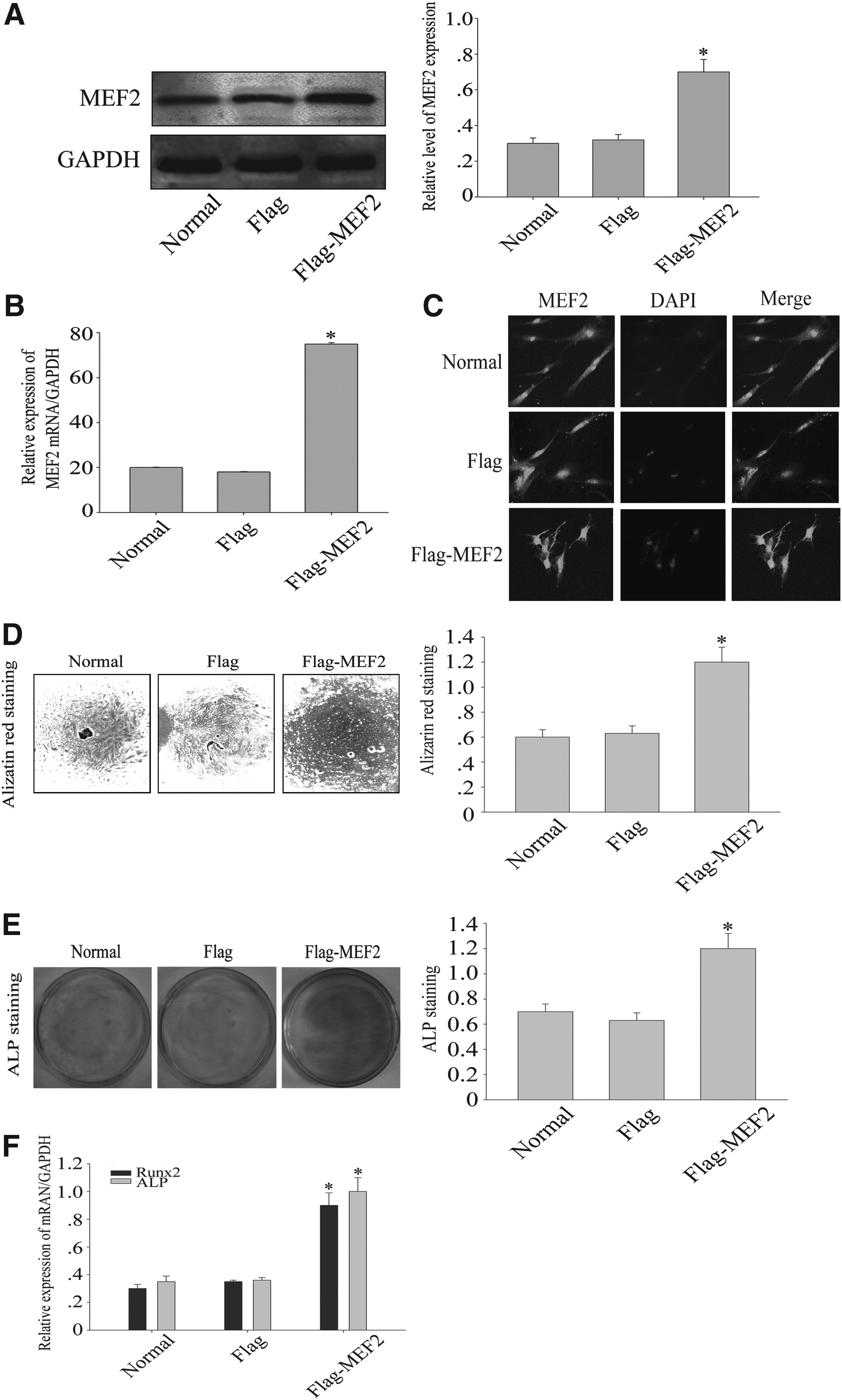
MEF2 overexpression in DPSCs by stable transduction. A full-length human MEF2 cDNA was transfected into DPSCs.
Inhibition of MEF2 inhibits osteogenesis in DPSCs
To further investigate the function of AGS3 during osteogenic differentiation of DPSCs, we built MEF2 knockdown siRNAs and transfection of pCMV-Flag-MEF2. As shown by Western blot and RT-PCR, siMEF2 had an effect on reducing MEF2 expression (Fig. 3A–C) (*p < 0.05). Then, ALP activity and Alizarin red S staining served to evaluate the effect of siMEF2 on osteogenic differentiation of DPSCs. As shown in Figure 3, mineralized matrix was strongly decreased on day 14 in the MEF2 silence DPSCs under osteogenic differentiation and the ALP activity was also decreased in comparison with the normal and siCon group (Fig. 3D–F) (*p < 0.05). Furthermore, RT-PCR showed that infection with siRNA resulted in a marked drop of ALP and Runx2 mRNA expression (Fig. 3G) (*p < 0.05). The above results showed that genetic silencing of MEF2 expression could inhibit osteogenic differentiation of DPSCs.

Inhibition of MEF2 decreased osteogenic differentiation of DPSCs.
MEF2 overexpression inhibits DPSC proliferation
To examine the effect of MEF2 on cell proliferation, MTT assay was used to reveal the proliferation of DPSCs. DPSCs were stably transfected with either pCMV-Flag vector or pCMV-Flag-MEF2. As show in Figure 4, MEF2 overexpression restricted the proliferation of DPSCs as determined by both cell counting (Fig. 4A) and MTT assay (Fig. 4B) (*p < 0.05).
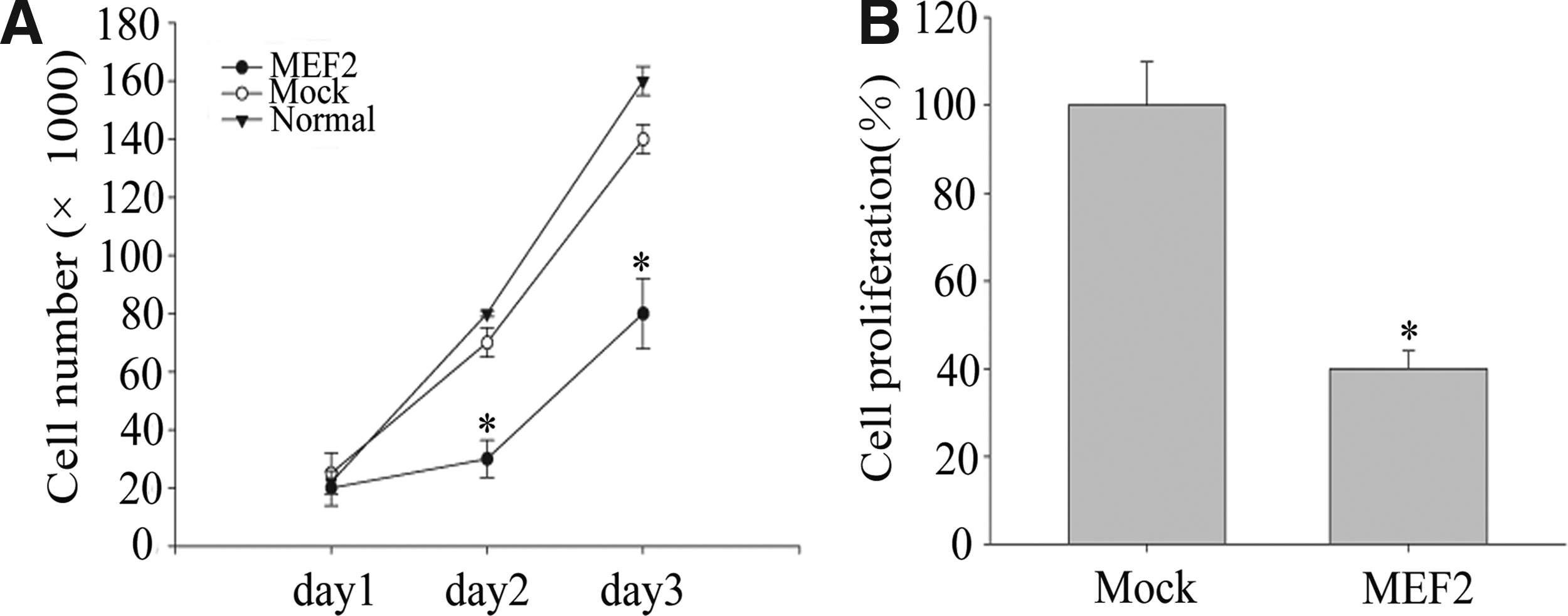
MEF2 overexpression decreased DPSC proliferation.
The effect of p38-MEF2 signaling on DPSCs
To examine the expression of p38-MEF2 signaling on DPSC osteogenic differentiation, we examined the expression levels of p38, p-p38, and MEF2. We found that SB202190, an inhibitor of p38, can inhibit p38 and p-p38 expression; moreover, MEF2 expression was significantly decreased. When DPSCs were overexpressed with MEF2 and treated with SB202190, we found that the expression of MEF2 was slightly higher, but still lower than that in the control group (Fig. 5A, B) (*p < 0.05). Next, osteogenic differentiation markers, ALP and Runx2, were detected. Western blot and mRNA expression showed that with the treatment of SB202190, the expression of ALP and Runx2 was decreased (Fig. 5C, D) (*p < 0.05). With MEF2 overexpression, we observed an enhancement in ALP activity and staining compared with the control group after 7 days of culture in osteogenic medium; however, this enhancement was reversed by p38 inhibitor SB202190 (Fig. 5E, F) (*p < 0.05). These findings support that inhibition of p38 activity could reverse the increase in osteogenic differentiation ability of MEF2 overexpressed DPSCs.

The effect of p38-MEF2 signaling pathway on DPSCs.
Discussion
The present work first studied the MEF2 role in DPSCs. Because MEF2 is mentioned in nerves and skeletal muscles that can control its differentiation, we examined whether MEF2 in DPSCs would affect their osteogenic differentiation. We observed that MEF2 increased in a time-dependent manner when DPSCs were under osteogenic differentiation medium, and MEF2 overexpression could inhibit proliferation of DPSCs. Moreover, blockade of MEF2 through the p38MAPK/MEF2 signaling pathway inhibitor reduced differentiation of DPSCs.
MEF2 is a specific transcription factor and is fascinating due to its involvement in diverse functions of gene regulation and multiplicity of regulatory mechanisms. The apparent ability of MEF2 is to control the transcription of genes in muscle differentiation (Naya and Olson, 1999). In this study, we found that MEF2 is required for osteogenic differentiation and mineralization of DPSCs. To better understand our work, this is the first time we investigated the role of MEF2 in osteogenic differentiation of DPSCs.
Recently, MEF2 has been shown to play a pivotal role in morphogenesis and myogenesis of skeletal, cardiac, and smooth muscle cells and bone (Olson et al., 1995). In skeletal muscle cells in culture, MEF2 has been reported to be expressed in proliferating myoblasts before the onset of differentiation (Breitbart et al., 1993). MEF2 is expressed in many cells, including muscle, neurons, and chondrocytes, accompanied by an activated differentiation program (Arnold et al., 2007; Verzi et al., 2007). MEF2 is a transcription factor that correlates with neuronal maintenance, survival, and differentiation (Heidenreich, and Linseman, 2004). Besides, MEF2C is the main transcription factor responsible for ECR5-dependent Sost transcription activation, which mediates bone formation (Collette et al., 2012). However, so far, reports on how MEF2 mediates bone formation are limited. In the present study, we found that MEF2 expression was low in early differentiation of DPSCs, but increased gradually when DPSCs were stimulated to differentiation. MEF2 stable expression led to enhanced ALP and Runx2 expression and activity in DPSCs and increased the mineralization of DPSCs.
The canonical interpretation of the p38MAPK-MEF2 signaling pathway has been modified by p38/MAPK-mediated phosphorylation, MEF2 transcriptional activation properties are enhanced (Han et al., 1997). Studies have demonstrated that MAP-kinase signaling controls several steps in the osteogenic pathway. Rapid stimulation of GPR30 by prunetin results in the production of cAMP and activation of Erk/MAPK that subsequently led to upregulation of Runx2, followed by GPR30 proteins in osteoblasts. Stimulation of this osteogenic signaling resulted in enhanced bone regeneration at the fracture site by prunetin (Khan et al., 2015). The ERK1/2-MAPK signaling pathway participates in IMS-induced differentiation of OVX BMSCs toward the osteogenic phenotype and plays a positive role in mechanotransduction (Zhang et al., 2015). Our study demonstrated that the p38MAPK/MEF2 pathway plays a key role in osteogenic differentiation of DPSCs. In our study, when MEF2 overexpressed, DPSCs were stimulated to differentiation and resulted in a dramatic increase in calcium deposition when compared with the control. When investigated, if interfered MEF2 expression could cause osteogenic differentiation, DPSCs were cultured under normal conditions for 7 days. Treatment with SB202190, an inhibitor of p38, could inhibit MEF2 expression. At the same time, ALP and Runx2 activities also decreased with the treatment of SB202190.
MAP kinase signaling has been shown to enhance the transcriptional activity of MEF2 factors in a variety of cell types. Expression and function of MEF2 transcription factors are controlled by diverse upstream regulatory systems. MAPKs, including p38 MAPKs, are major HDAC-independent regulators of MEF2. p38 MAPKs phosphorylate MEF2 to augment its function in myoblasts (Zetser et al., 1999), monocyte/macrophage-like cells (Han et al., 1997), and so on. The p38 pathway plays an essential role in the differentiation of myoblasts and this process may be mediated through the MEF2 transcription factor (Zetser et al., 1999). In primary chondrocytes, p38 MAPK inhibition reduces MEF2C function and hypertrophic differentiation (Stanton et al., 2004). In a previous study, the enhancement is mediated by phosphorylation of three amino acids in the C-terminal activation domain of MEF2 by the MAP kinase family member, p38 (Han et al., 1997). Moreover, phosphorylation of a component can be implemented by different kinases, which can phosphorylate MEF2 family members through different phosphorylation sites. MAP kinase members dramatically increase the transactivation activity of MEF2 by phosphorylating a serine residue at amino and the phosphorylation of MEF2 results in increased transcriptional activity (Kato et al., 1997).
Previous study also showed that in 293 cells, phosphorylation of MEF2 by p38 enhanced MEF2-dependent gene expression (Zhao et al., 1999). This indicated the possible regulatory pathways that depend on p38-mediated phosphorylation of transcription of MEF2. In our study, p38 was probably the kinase that phosphorylated MEF2 because p38 activity was significantly inhibited in DPSCs treated with SB202190, and if cells were treated with SB 203580 during that period, the induction of MEF2 expression was significantly abolished (Fig. 5A, B). In other reports, p38 MAPK signaling and inhibition of p38 reduced MEF2 expression to inhibit the hypertrophic differentiation program (Papaioannou et al., 2015). Above all, p38 MAPK is a potential activator of MEF2 and it is expected to control osteogenesis differentiation of DPSCs.
In conclusion, the present study suggests that in regulating DPSC proliferation and osteoblast differentiation, MEF2 plays a previously unknown role. We have demonstrated that MEF2 is expressed in DPSCs, which is a novel finding. We show by in vitro experiments that MEF2 modulates the function of DPSCs partially by p38/MEF2 signaling. In brief, MEF2 is not only an inhibitor of DPSC proliferation but also a promoter of DPSC osteogenic differentiation. Future studies will be necessary to investigate additional experiments to address the mechanism of MEF2 on osteogenic differentiation of DPSCs.
Footnotes
Acknowledgments
This work is supported by the National Natural Science Foundation of China under Grant 81500809 and Funding of Jiangsu Innovation Program for Graduate Education KYZZ15-0091, the Fundamental Research Funds for the Central Universities; the Graduate Student Innovation of Science and Technology Projects in Jiangsu Province (Grant no. SJLX0588); and the Top Six Types of Talents Financial Assistance of Jiangsu Province Grant (No. 3013-WSN-076).
Author Disclosure Statement
The authors declare that no conflicting financial interests exist.