
Abstract
Abstract
Although animal cloning is becoming increasingly practicable, cloned embryos possess many abnormalities and so there has been a low success rate for producing live animals. This is most likely due to incomplete reprogramming of somatic cell nuclei before they start to develop as the donor nuclei are usually only exposed to the oocyte cytoplasm for 1–2 hours before reconstructed oocytes are activated to avoid oocyte aging. Therefore, in this study, we attempted to extend the exposure period of somatic cell nuclei to the oocyte cytoplasm to determine whether this could enhance reprogramming of donor nuclei. Donor nuclei were transferred into oocytes, following which pseudo-MII spindles (pMIIs) derived from these were extracted and injected into newly collected enucleated oocytes 24 hours after the first nuclear transfer (NT). These serial NT oocytes were then activated and their developmental potential was examined to full term. There was no obvious difference in the pMIIs of reconstructed oocytes at 6 and 24 hours after donor nucleus injection; however, in both of these, the chromosomes were more widely spread inside the spindle than in fresh intact oocytes. Furthermore, a few chromosomes remained in 25% and 47% of these enucleated oocytes at 6 and 24 hours after donor nucleus injection, respectively. When these pMIIs were injected into fresh enucleated oocytes, the developmental rate to blastocyst was significantly lower, but we could still obtain several healthy cloned offspring. Thus, serial NT at intervals of 24 hours using fresh oocytes is possible, but the success rate could not be improved due to loss of chromosomes during the second NT.
Introduction
T
However, many of the resulting cloned embryos/fetuses showed aberrant epigenetic methylation patterns in their DNA or acetylation patterns in their nuclear histones (Dean et al., 2001; Kang et al., 2001; Ohgane et al., 2001). Therefore, these studies concluded that the low success rate in successfully developing cloned embryos to full term and the large number of anomalies among cloned embryos may arise from incomplete epigenetic reprogramming of the donor nucleus. Supporting this, it was found that the cloning success rate was improved when the histone deacetylase inhibitor, trichostatin A (TSA), was used to correct epigenetic abnormalities (Kishigami et al., 2006a), and many other agents for controlling the epigenetic status have since been trialed in attempts to improve cloning efficiency (Inoue et al., 2010; Matoba et al., 2014; Ono et al., 2010; Thuan et al., 2010; Van Thuan et al., 2009). However, the generation of live offspring is still inefficient.
Serial NT is a method used to improve nuclear reprogramming. In the study that pioneered this approach, differentiated somatic cell nuclei of Xenopus were transplanted into oocytes or eggs, and developing cloned embryos were then used as a second nuclear donor. This study demonstrated that serial NT can improve the reprogramming and success rate when cloning frogs, but unfortunately this success rate remains low (DiBerardino and Hoffner, 1983; Gurdon et al., 1975).
In domestic animals, serial NT was originally used to increase the number of cloned embryos from single donor embryos because, at that time, it was not possible to produce cloned animals from somatic cell nuclei and there was a limitation to the number of donor cells that could be collected from a single embryo (Stice and Keefer, 1993; Westhusin et al., 1991). Unfortunately, however, the success rate of cloning cows decreased with an increase in the number of NT repeats. Once somatic cell cloning was achieved (Wilmut et al., 1997), this particular cloning limitation was solved and so it was thought that serial NT was a redundant technique. However, we have since demonstrated that serial NT is a useful technique in mice (Mizutani et al., 2008; Wakayama et al., 2005) and that it can be repeated at least 25 times without decreasing the success rate (Wakayama et al., 2013). However, an increased number of NT repeats did not increase the success rate to full term, demonstrating that serial NT could not improve the reprogramming of the donor nucleus using traditional NT techniques.
In the traditional NT method, donor nuclei are exposed to the oocyte cytoplasm for a few hours before oocyte activation (Campbell et al., 1996; Kishigami et al., 2006b). It is likely that the donor nuclei are reprogrammed during this period, but only a few reconstructed oocytes obtain the complete capacity for full-term development. Therefore, it is possible that if the exposure period of donor nuclei to the oocyte cytoplasm could be extended, the reprogramming competency of the donor nuclei may be increased. However, such an experiment has not been performed to date because reconstructed oocytes need to be activated within a few hours of NT to prevent them from aging, even if reprogramming is not yet complete.
In this study, we attempted to extend the exposure period of somatic cell nuclei to the oocyte cytoplasm using serial NT techniques. It has previously been shown that when oocytes are cultured in a 37°C incubator, none of the oocytes can maintain their developmental potential until the next day due to in vitro aging (Ono et al., 2011; Wakayama et al., 2004). Therefore, following exposure of donor nuclei to the oocyte cytoplasm for up to 24 hours, the donor nuclei were removed from reconstructed oocytes and transferred again into newly collected fresh enucleated oocytes. We then examined the developmental potential of these serial cloned embryos and chromosome loss using this technique.
Materials and Methods
Animals
We used 8–10-week-old B6D2F1 (C57BL/6 N × DBA/2) mice to produce oocytes and cumulus cells as well as female ICR strain mice that had been mated with vasectomized males of the same strain as surrogate pseudopregnant embryo transfer recipients. The B6D2F1 and ICR mice were purchased from the Shizuoka Laboratory Animal Center (Hamamatsu, Japan). All animal experiments conformed to the Guide for the Care and Use of Laboratory Animals and were approved by the Institutional Committee of Laboratory Animal Experimentation of the University of Yamanashi.
Collection of oocytes
The 8–10-week-old female mice were induced to superovulate with 5 IU pregnant mare serum gonadotropin (Teikokuzoki, Tokyo, Japan), followed by 5 IU human chorionic gonadotropin (hCG) (Teikokuzoki) 48 hours later. We collected cumulus–oocyte complexes from their oviducts ∼16 hours after hCG injection, and then placed these in HEPES-buffered CZB medium (H-CZB) (Chatot et al., 1990) and treated them with 0.1% bovine testicular hyaluronidase (Sigma-Aldrich, St. Louis, MO). After several minutes, we washed the cumulus-free oocytes twice and then placed them in a droplet of CZB medium for culture.
First round of NT
NT was performed as previously described (Kishigami et al., 2006b; Wakayama et al., 1998). Briefly, we treated the unfertilized oocytes with CB for 10 minutes and then removed the MII spindles (hereafter, MIIs) with an enucleation pipette (7–8 inner diameter). Enucleated oocytes were injected individually with these cumulus cell nuclei. Following NT, the reconstructed oocytes (hereafter, the first NT oocytes) were kept in CZB medium in a 37°C, 5% CO2, incubator until the morning of the next day (Fig. 1). As a control, we activated the reconstructed oocytes using 5 mM SrCl2 in Ca2+-free CZB medium in the presence of 5 μM Lat A supplemented with 50 nM TSA for 9 hours (Terashita et al., 2012). It is important to note that in this study, lysis of many of the oocytes occurred during the activation process. The reason for this was not known. We therefore used 5 mM SrCl2 rather than 10 mM, which allowed activation of most of the oocytes without lysis. After three washes in CZB, the cloned embryos were cultured in the same medium for in vitro development or until embryo transfer into a recipient female.
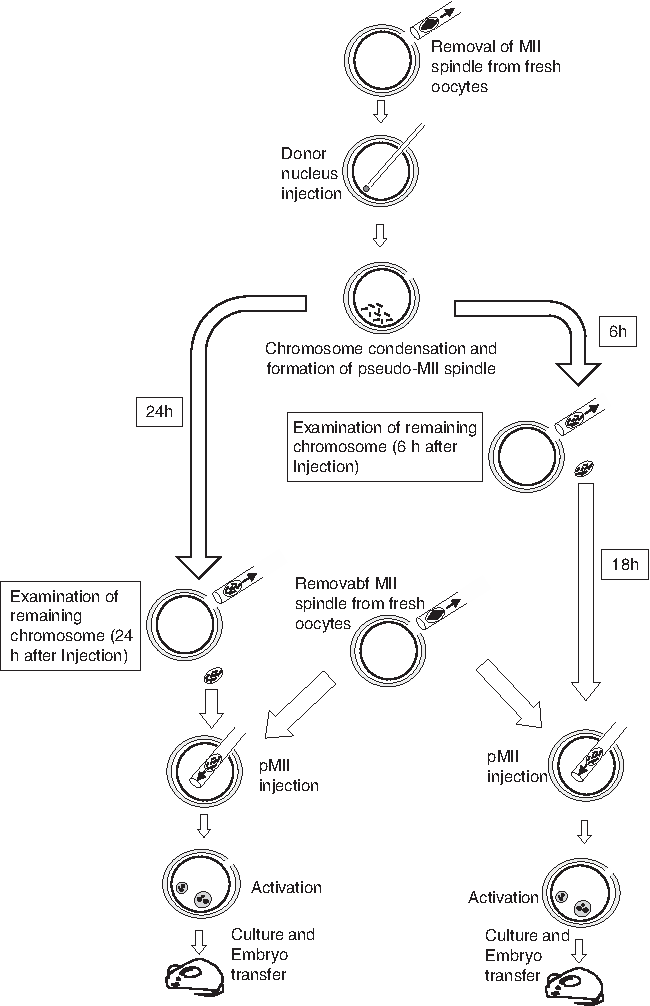
Schematic diagram of the serial NT procedure. Donor nuclei were injected into fresh enucleated oocytes and exposed to the oocyte cytoplasm until the following day; a second NT was performed using fresh enucleated oocytes. NT, nuclear transfer.
Second round of NT
At either 6 or 24 hours after NT, we treated the first NT oocytes with CB, and then removed the MII-like structure (pseudo-MII spindle; hereafter pMIIs) derived from the cumulus cell nuclei. When the pMIIs were collected at 6 hours after NT, the karyoplasts were kept in CZB medium in a 37°C, 5% CO2, incubator until they reached 24 hours (Fig. 1). The next day, before the start of the second round of NT, we enucleated newly collected fresh oocytes in the usual way. We then injected the pMIIs into the enucleated oocytes (hereafter, second NT oocytes). These second NT oocytes were placed in CZB medium for 2–3 hours and then activated as usual. Since this time matched that of the control donor cell nuclei, the cumulus cells that had been preserved in PVP medium for 24 hours at room temperature were also injected into fresh oocytes and activated.
Observation of first NT oocytes at 6 or 24 hours after NT
We fixed the first NT oocytes in 4% paraformaldehyde in phosphate-buffered saline at either 6 or 24 hours after NT. Following permeabilization and blocking, these oocytes were incubated with mouse monoclonal anti-β-tubulin (1:100 dilution; Anti-beta Tubulin antibody labeled with FITC, ab11310; Abcam) overnight at 4°C. We then stained the DNA of the oocytes with propidium iodide (5 μg/mL) and observed them using a confocal scanning laser microscope (FV-1200; Olympus).
Examination of remaining chromosomes in enucleated first NT oocytes
To assess whether all of the pMIIs had been removed from the first NT oocytes, we double stained the enucleated first NT oocytes. For histone staining, we used anti-H3S10ph antibodies labeled with phycoerythrin (Cell Signaling Technology, Beverley, MA) at 1/100 dilution. The antibody solution was aspirated into a pipette with a micromanipulator attached, following which a few picoliters of solution were introduced into the first NT oocytes, as described previously (Yamagata et al., 2012). For DNA staining, these oocytes were introduced into H-CZB medium containing 5 μg/mL Hoechst 33342 for 1 minute. We examined the double-stained oocytes by fluorescent microcopy and counted the number of chromosomes remaining in each enucleated oocyte.
Developmental potential of second NT oocytes following activation
The cloned embryos were cultured until 72 hours after activation, during which time we examined the developmental rates to the blastocyst stage. To produce offspring, we transferred two-cell stage embryos or morulae/blastocysts into the oviducts or uteri, respectively, of pseudopregnant ICR females. Even though there were only a small number of developed cloned embryos at the time of embryo transfer, we did not mix them with any supporting (fertilized) embryos in this study.
Cloned offspring were then obtained at 19.5 dpc by cesarean section.
Statistical analysis
We evaluated offspring development rates using chi-squared tests, with a significance level of p < 0.05.
Results
Observation of pMIIs in oocytes at 6 or 24 hours after NT
When the pMIIs of first NT oocytes were observed at 6 or 24 hours after NT, the spindles were elongated and the chromosomes were mostly spread inside the spindle, although, in some cases, a few chromosomes were also detected outside the spindle (Fig. 1A–C). There was no obvious difference between 6- and 24-hour-old first NT oocytes (Fig. 2B, C), but both were very different from fresh oocytes, which exhibited compact spindles and lined up chromosomes (Fig. 2A). When pMIIs were removed from the first NT oocytes, it was somewhat difficult to detect the location of the spindle compared with the MIIs of fresh oocytes. However, most of the pMIIs could be removed from all of the first NT oocytes.
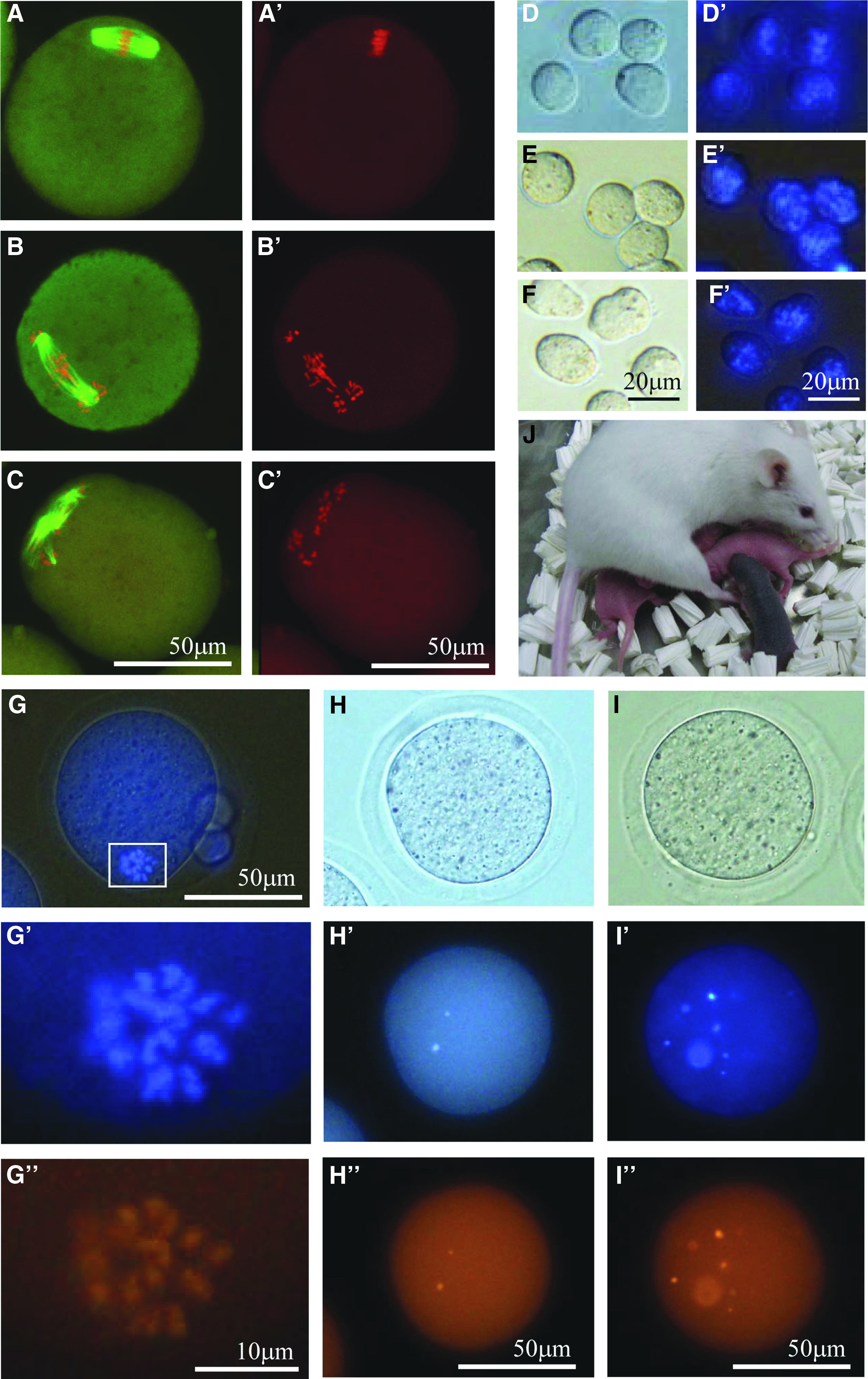
Aberrant spindle formation and detection of failed enucleation in aged cloned oocytes and production of cloned mouse.
All of the karyoplasts collected from first NT oocytes at 6 or 24 hours after NT showed scattered chromosomes inside the cytoplasm, whereas most of the MII karyoplasts collected from fresh oocytes were compact with their chromosomes lined up inside the cytoplasm (Fig. 2E, F). These results may suggest that we failed to remove all of the chromosomes from the first NT oocytes during the enucleation process.
Observation of enucleated first NT oocytes at 6 or 24 hours after NT
To examine whether enucleation of pMIIs was successful, we double stained the enucleated oocytes (Fig. 2G–I). When the first NT oocytes were enucleated at 24 hours after NT, 47% possessed a few chromosomes inside the oocyte cytoplasm, with five of the enucleated oocytes possessing more than six chromosomes each (Table 1). By contrast, when enucleation was performed at 6 hours after NT, only 25% of the oocytes possessed a few chromosomes. In the control, where MIIs were removed from fresh oocytes, none of the enucleated oocytes contained chromosomes. Therefore, in the following experiment, we removed the pMIIs from oocytes at 6 hours after NT, and then kept those karyoplasts in an incubator until the next day, at which stage the pMIIs were transferred into fresh enucleated oocytes.
Superscript “a” versus “b” indicates a significant difference (p < 0.05).
pMIIs, pseudo-MII spindles; NT, nuclear transfer.
Developmental potential of second NT oocytes following activation
When the pMIIs were collected from the first NT oocytes, relatively few of the cloned embryos developed to the morula/blastocyst stage (18% at 6 hours and 10% at 24 hours after first NT; Table 2). These levels were significantly lower than the control (85%) or time-matched control (87%). Following embryo transfer into recipient females, only one offspring (Fig. 3D; black-eyed pup) was obtained from the cloned morulae/blastocysts, which was from a pMII that had been collected at 6 hours after NT.
Superscript “a” versus “b” indicates significant differences (p < 0.05).
Two-cell embryos were transferred into recipient female.
Cont, control; ET, embryo transfer; Frag., fragmentation; Moru/blast, morula/blastocyst; PN, pronuclear stage.
Serial cloned embryos were transferred to the recipient females at the two-cell stage to avoid the negative effect of a long period of in vitro culture. We did not use the 24-hour-old first NT oocytes in this experiment due to their very low developmental success rate. The developmental rate to the two-cell stage was 94% and the production rate of cloned offspring was 2%, which was similar to the control and time-matched control. These clones grew normally to adulthood and demonstrated their fertility after being mated with wild-type male mice.
Discussion
In traditional NT, reconstructed oocytes are usually activated within 1–2 hours of NT and the donor nuclei are reprogrammed during this period. Therefore, it is possible that reprogramming of donor nuclei and the developmental potential will be enhanced if donor nuclei are exposed to the oocyte cytoplasm for a longer period. However, it is also well known that oocytes have a very short life span and quickly lose their developmental potential (Ono et al., 2011; Wakayama et al., 2004). This suggests that even if donor nuclei can be reprogrammed by a prolonged exposure to the oocyte cytoplasm, these reconstructed oocytes will be aged and thus lose their developmental potential. Therefore, no previous studies have investigated the relationship between the exposure period of the donor nuclei to the cytoplasm of oocytes and the completeness of reprogramming. In this study, we resolved the issue of in vitro aging of the reconstructed oocytes by establishing a serial NT procedure, in which the donor nuclei of reconstructed aged oocytes were taken and injected into newly collected fresh enucleated oocytes.
A previous study of in vitro aging of mouse oocytes showed that the size of the spindle increases with increased oocyte aging (Ono et al., 2011). Thus, we first observed the size of pMIIs at 6 or 24 hours after NT. This showed that the size of pMIIs had already expanded at 6 hours after NT, but did not expand further when cultured for up to 24 hours. Therefore, we thought that it would be possible to enucleate the pMIIs even at 24 hours after NT. However, following enucleation, a few chromosomes remained in nearly half of the 24-hour-old first NT oocytes, demonstrating that enucleation had failed. By contrast, when enucleation was performed at 6 hours after NT, significantly fewer enucleated oocytes contained chromosomes. Previously, we reported that 24-hour-old NT oocytes displayed more severely scattered chromosomes or irregularly shaped spindles (Lee et al., 2016). These may suggest that the tension of the tubulin was weaker in the 24-hour-old spindles than 6-hour-old spindles, despite having the same size, resulting in some of the chromosomes failing to be sucked up by the enucleation pipette.
During the additional culture of the 6-hour-old pMIIs (which were kept in vitro until the next day), the small karyoplasts may have been negatively affected, and the final developmental competence may have been compromised. It would have been preferable to use another batch of mice that were treated with hormones 6 hours later (e.g., from another light–dark controlled room) as a fresh MII oocyte source for the second NT. If this had been possible, the success rate of mouse cloning would probably have been higher than after a 24-hour interval.
When these defective karyoplasts were used as donor for serial NT, the reconstructed cloned oocytes also lacked a few chromosomes and the defective cloned embryos could not develop to full term. Therefore, unfortunately, we were unable to clarify whether prolonged exposure of donor nuclei to the oocyte cytoplasm enhanced nuclear reprogramming. To improve the serial NT procedure, it is important to prevent oocyte aging during reprogramming, such as by using caffeine or MG132 during oocyte culture, which may prevent the spindles from aging (Josefsberg et al., 2000; Kikuchi et al., 2002), or simply, increase the concentration of NaCl in the culture medium; in this study, we observed disturbed microtubules, irregular spindles, and scattered chromosomes, which were also observed in rat cloning when MAPK activity was decreased in aged oocytes. Interestingly, those phenomena were corrected and MAPK activity was increased when the concentration of Na+ in the culture medium was increased (Cui et al., 2012, 2013).
This serial NT methodology may also be useful for other basic studies. For example, the effect of donor cytoplasm on cloning is still unclear. A small volume of donor cytoplasm is usually transferred into the recipient oocyte at the same time as NT, resulting in donor mitochondria often being detected in cloned animals (Evans et al., 1999; Inoue et al., 2004). It is not currently known whether this transferred donor cytoplasm causes negative effects on the development of cloned embryos. However, the use of the serial NT procedure will result in the donor cytoplasm being diluted in the first recipient oocyte and so very little donor cytoplasm will be transferred into the second recipient oocyte. Previously, we reported that somatic cell cytoplasm affected placental development when it was injected into fertilized embryos (Van Thuan et al., 2006). However, interestingly, one of the cloned mice in the present study was born naturally, which is very unusual as nearly all cloned mice need to be delivered by cesarean section due to the lack of natural delivery in pregnant recipient females. Therefore, these findings suggest that it may be the cytoplasm of the donor cells that affects the placental abnormality (Wakayama and Yanagimachi, 1999) and thus the lack of natural delivery.
In conclusion, although we failed to increase the success rate of mouse cloning using serial NT in this study due to chromosome loss, once this issue has been resolved, this technique will provide a new and unique tool to examine various questions around animal cloning.
Footnotes
Acknowledgments
The authors thank Miss. K. Kishida for critical comments on the study. This work was partially funded by the Japan Society for the Promotion of Science to E.M. (15H04605), S.K. (26450458), and T.W. (23248048 and 16H02593); the Naito Foundation to S.W.; PRESTO of the Japan Science and Technology Agency to S.K.; Asada Science Foundation to T.W.; and the Takeda Science Foundation to T.W.
Author Disclosure Statement
The authors declare that no conflicting financial interests exist.