
Abstract
Abstract
Emerging evidence suggests that epitranscriptional modifications influence multiple cellular processes. N6-methyladenosine (m6A), as the most abundant reversible methylation of mRNA, has also been reported to play critical roles in modulating embryonic stem cell differentiation and somatic cell reprogramming by regulating gene expression. This review examined the characteristics of m6A, including the distribution profile and currently discovered “writer,” “eraser,” and “reader” proteins. Moreover, the hypothesis is proposed that m6A could influence cell fate determination, and the underlying mechanisms are due to the related mRNA degradation, causing weakening of previous cell characteristics and eventually leading them to develop into the reverse direction (pluripotency or differentiation state). Accordingly, m6A modifications presented its potential role in cell fate determination, which provides new insights into understanding the mechanisms of various diseases.
Introduction
R
The distribution profile of m6A in transcripts shows it is significantly enriched in 3′ untranslated regions (3′UTRs) and near the stop codon. m6A sites also appear in a sequence feature (Bodi et al., 2010; Dominissini et al., 2012; Meyer et al., 2012) and they seem to be restricted to a consensus multiple RRACH (where R denotes A or G, and H denotes A, C, or U) motif. However, m6A can also exist at other nonconsensus sites (Csepany et al., 1990; Harper et al., 1990; Meyer and Jaffrey, 2014; Wei and Moss, 1977).
In vivo, the formation of m6A is a reversible biological process. The dynamic methylation of m6A is regulated by “writer” and “eraser” proteins (Fig. 1A). In eukaryotes, m6A “writers” were identified as the methyltransferase (MTase) complex consisting of three individual components: methyltransferase-like 3 (METTL3), methyltransferase-like 14 (METTL14), and Wilms' tumor 1-associating protein (WTAP) (Bokar et al., 1997; Finkel and Groner, 1983; Schwartz et al., 2014).
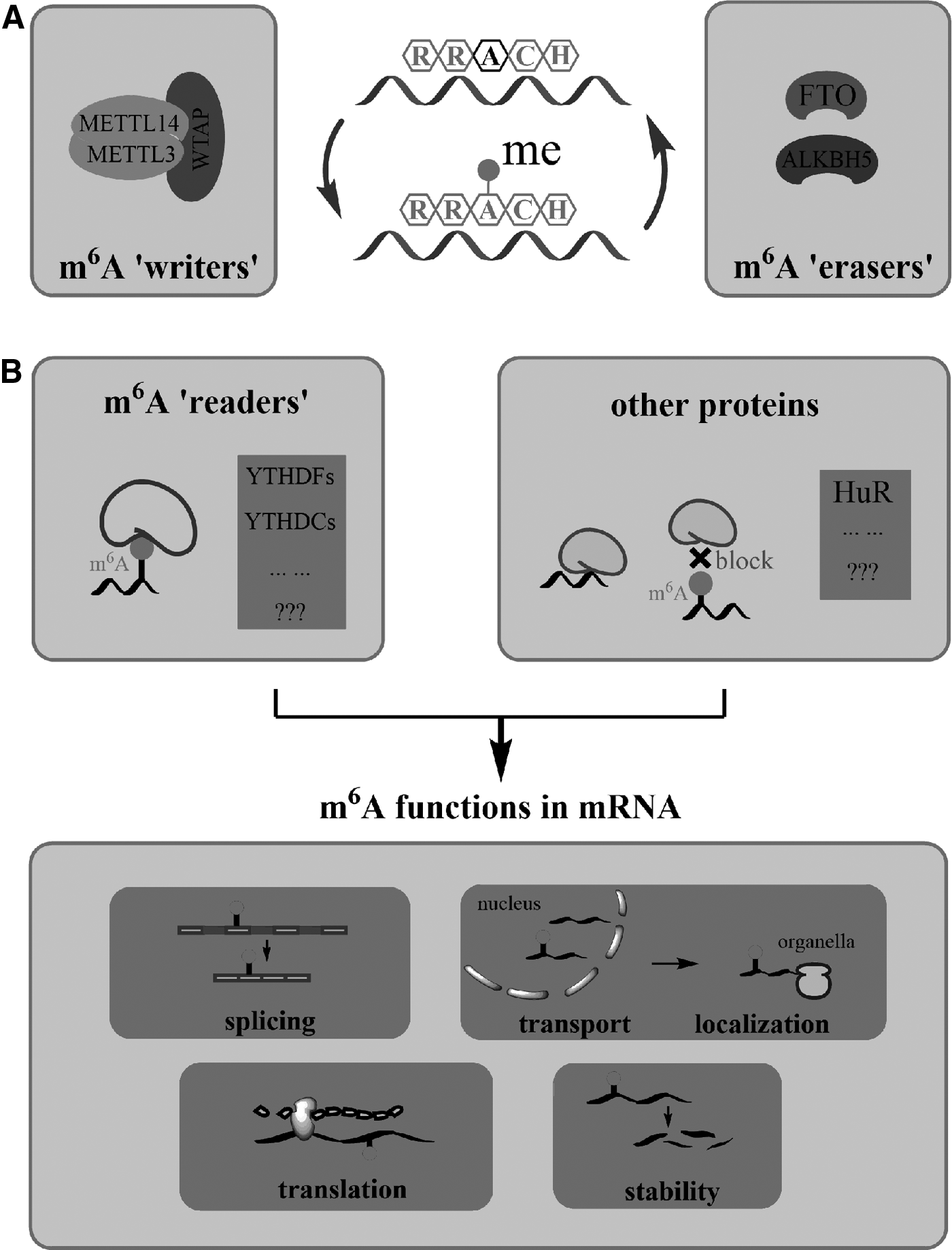
Reversible m6A modification and its functions in mRNA.
A related study in mouse embryonic stem cells (mESCs) also indicated that METTL3 and METTL14 work as a complex (Wang et al., 2014). Although both of them have the methylation activity, when they work as a heterocomplex, the methylation ability will be substantially improved (Wang et al., 2016). Liu et al. (2014) reported that knockdown of WTAP leads to a decrease in the total m6A level in HeLa and 293FT cells. WTAP interacts with both METTL3 and METTL14 and colocalizes with the METTL3–METTL14 heterodimer in nuclear speckles to participate in m6A RNA methylation (Horiuchi et al., 2013). These indicate that WTAP has important roles in cellular m6A deposition (Little et al., 2000; Ping et al., 2014).
There is evidence that the complete m6A methylation capability in mammals also requires KIAA1429, and its Drosophila homolog (virilizer) is crucial for sex determination by altering splicing of sex determination factor Sex-lethal (Sxl) (Haussmann et al., 2016; Lence et al., 2016). Besides, one recent research showed that RNA-binding motif protein 15 (RBM15) and its paralogue RBM15B could recruit the m6A MTase complex to specific sites and influence m6A formation in mRNA and a specific long noncoding RNA (lncRNA) (Patil et al., 2016). Up to now, two kinds of m6A “erasers” have been discovered to show demethylation activity: fat mass and obesity protein (FTO) (Jia et al., 2011) and AlkB homolog 5 (ALKBH5) (Zheng et al., 2012). Both of these AlkB family proteins belong to the nonheme Fe (II)/α-ketoglutarate (α-KG)-dependent dioxygenase.
As a reversible mRNA modification, previous studies have shown that m6A could affect mRNA stability, splicing, transport, translation, and localization, and further influence gene expression leading to various biological processes (Fig. 1B) (Camper et al., 1984; Fustin et al., 2013; Hess et al., 2013; Shah and Clancy 1992; Tuck et al., 1999; Zhao et al., 2014). Researches also showed that m6A is related to some human diseases, such as obesity and cancer (Dominissini et al., 2012; Jia et al., 2011; Machnicka et al., 2013; Meyer et al., 2012; Sibbritt et al., 2013). m6A modification could be read by some “reader” proteins to exert its biological functions.
For example, YTHDF2 (one of YTH domain family located in the cytoplasmic region) can specifically bind to m6A-containing mRNAs and could mediate degradation of these mRNAs (Kang et al., 2014; Wang et al., 2014; Xu et al., 2014; Zhu et al., 2014). YTHDC1 (one of YTH domain family located in the nuclear region) can regulate mRNA splicing. It interacts with splicing factor SRSF3 and enhances SRSF3s binding to mRNA exons via m6A, which eventually promotes exon inclusion and, at the same time, YTHDC1 prevents SRSF10 binding to mRNA and obstructs exon skipping (Roundtree and He, 2016; Xiao et al., 2016). Besides, heterogeneous nuclear ribonucleoprotein could be another potential m6A reader and is related to localization and transportation of mRNA (Dominissini et al., 2012). Furthermore, the presence of m6A could also block the binding of some proteins to m6A-modified sites or alter the high-level structure of mRNA to sequentially affect protein-RNA interactions.
In general, m6A readers could recognize m6A-marked mRNAs, change their transport and localization, and then influence mRNA stability, splicing, or translation, and finally, affect gene expression through posttranslational regulation. However, the regulatory mechanisms and exact functions of m6A still remain poorly understood.
m6A Influences mRNA Degradation
m6A has been suggested for its several functions. It is involved in different cellular processes, of which regulating the stability of mRNA is an outstanding feature (Ingolia et al., 2011). Previous studies have shown that m6A modification-enriched mRNA presented decreased stable states compared to the less modified ones. In the research by Wang et al., m6A depletion after knockout of the methyltransferase METTL3 and METTL14 in embryonic stem cells (ESCs) results in reduction of mRNA decay rates along with the prolonged half-life (Wang et al., 2014). Thereby, the translation efficiency of m6A-modified transcripts was obviously lower than unmodified ones. All these findings indicate that the presence of m6A reduces the stability and promotes the degradation of the mRNA.
m6A modification destabilizing mRNA is associated with its readers, YTH domain family members 1, 2, and 3 (YTHDF1-3), among which YTHDF2 presents higher selectivity to m6A (Li et al., 2014; Zhu et al., 2014). According to the study by Fu et al., YTHDF2 showed 16-fold higher binding affinity to m6A-containing mRNAs (preferably in a conserved Gm6AC motif of mRNAs) compared to the unmethylated ones (Fu et al., 2014). Besides, this binding induced the YTHDF2-mRNA complex to relocalize from actively translating pool to P-bodies, which are the cellular RNA decay sites. m6A thereby results in accelerated mRNA decay, shortening the lifetime of mRNA.
Another study also approved m6A destabilized mRNAs, however, by interfering with the HuR-microRNA pathway (Srikantan et al., 2012). The HuR-mRNA complex can prevent miRNA-mediated mRNA decay. This study showed that m6A modification inhibited the binding ability of human antigen R (HuR, an RNA-binding protein that can increase RNA stability) to mRNA, therefore accelerating the mRNA decay. Certainly, METTL3/14 knockdown cells showed increased HuR-mRNA binding accompanied with prolonged mRNA lifetime compared to control, which revealed that existence of m6A blocks HuR-mRNA binding and further promotes the destabilization of mRNA (Kundu et al., 2012; Lebedeva et al., 2011; Srikantan et al., 2012; Wang et al., 2014). Although all these studies indicated that m6A modification affects the status of mRNA, the underlying mechanisms are still not clear and need to be further investigated.
m6A Regulates ESC State
As we mentioned above, m6A affects the stability of mRNA and further influences the expression of some proteins. Consequently, the different level of m6A might play an important role in numerous biological processes. For instance, numerous master regulators of ESC maintenance and differentiation contain m6A modifications, including Nanog, Sox2, Nr5a2, Eomes, and FoxA2 (Dunn et al., 2014; Young, 2011). Indeed, research has established that m6A is essential for cell differentiation and development in mammals.
In 2014, Batista et al. (2014) observed that METTL3 knockout ESC exhibits the decreased level of apoptosis and a promoted proliferation rate compared with wild-type ESC. METTL3 knockout ESC presented lower levels of m6A and further increases the expression of master genes, resulting in the promotion of the ESC self-renewal, maintenance of the state of pluripotency, and hinders differentiation. These observations suggested that m6A has a significant role in regulating ESC state: m6A deficiency assists ESC to maintain pluripotency and resist differentiation.
Closely behind, Aguilo et al. (2015) also showed that lower m6A levels in ESC-related transcripts enable pluripotency and renewal. This study demonstrates that the chromatin-associated zinc finger protein 217 (ZFP217) is critical for the maintenance of ESC self-renewal and somatic cell (SC) reprogramming. It regulates the transcription of pluripotency genes and prevents such transcription from aberrant m6A methylation. However, another study reported by Wang et al. in 2014 showed an opposite result that the decreased cell proliferation rate was detected in both METTL3 and METTL14 knockdown mESCs (Wang et al., 2014). In the meanwhile, pluripotency factors were downregulated, while developmental regulators were upregulated. This study suggests that m6A deficiency reduces stem cell features of mESCs instead.
These inconsistent results can be explained combining with a latest study (Geula et al., 2015; Zhao and He, 2015), which suggested the stem cell includes two distinct states: naive (ESC) state and primed pluripotent (epiblast stem cell) state. Most transcripts of both pluripotency genes and lineage-commitment genes contain m6A modifications. However, in the naive state and primed pluripotent state, naive pluripotency genes and lineage-commitment genes are dominant, respectively. Thus, we can understand the regulation effects of m6A in ESC state from two respects. ESCs in naive state presented maintenance of pluripotency state after knockout of the METTL3.
The reason that in the naive state demethylation of m6A increases the stability of methylated transcripts and further upregulates the already high naive pluripotency genes to create a “hyper”-naive pluripotency state is because cells tend to maintain pluripotency at the ESC state. On the contrary, demethylation during the primed state boosts the dominating lineage-commitment factors and tips the balance toward differentiation.
In conclusion, m6A RNA methylation dynamically regulated the ESC state that m6A deficiency can either encourage the maintenance of the pluripotency or promote the differentiation, respectively, depending on whether ESC is in the naive or primed pluripotent state. Batista and Aguilo studied in the naive state of ESC and got the conclusion that m6A could boost differentiation, while Wang studied in the primed pluripotent state and obtained a distinct result. Anyhow, no matter which state the ESC is in, m6A modification is critical in regulating ESC state.
m6A Promotes Reprogramming
m6A not only influences the fate of ESCs but also plays a significant role in controlling SC reprogramming. A recent article reported by Chen et al. (2015) revealed that increased m6A formation promotes cell reprogramming to pluripotency. Transcripts of many cell-type-specific marker genes contain m6A modification, such as Nanog, POU3F2, and DHH. These genes involve in cell fate determination. To identify whether m6A affects cell fate, they overexpressed human Myc-METTL3 in mouse embryonic fibroblasts (MEFs) transduced with four Yamanaka factors (Sox2, Klf4, Oct4, c-Myc; SKOM). These MEFs presented a raised level of m6A, increased colonies of induced pluripotent stem cell (iPSC), and enhanced expressions of key pluripotent factors (Oct4, Sox2, and Nanog) during the reprogramming process. Conversely, knockdown METTL3 can result in iPSC colony reduction and pluripotent gene expression decline. In addition, they overexpressed human Myc-METTL3 into METTL3 knockdown MEFs, which partly rescued the inhibited reprogramming efficiency. These data give evidence on the m6A promoting cell reprogramming process.
On the contrary, Aguilo et al. seemed to indicate that the increment of m6A levels occurring on Zfp217 knockdown maybe a barrier for efficient SC reprogramming (Aguilo et al., 2015). They performed reprogramming in Zfp217 knockdown MEFs, and the depletion of ZFP217 increased the level of m6A, and meanwhile, lowered the expressions of 3/4 Yamanaka factors: Sox2, Klf4, and c-Myc. However, in normal MEFs, increased m6A could raise expressions of some pluripotent genes (Oct4, Sox2, and Nanog) (Chen et al., 2015). This is more convincing that the impaired SC reprogramming was caused by loss of ZFP217 rather than directly due to the rise of the m6A level. Collectively, m6A increased directly by the expression change of methyltransferase could promote SC reprogramming.
Chen's group also demonstrated that m6A methylation was regulated by microRNAs (miRNAs). They found that a large proportion of m6A sequence motifs could pair with miRNAs. A positive correlation was observed between the level of m6A and Dicer, an essential endonuclease for producing mature miRNAs. Knocking down Dicer significantly reduced cellular m6A abundance, while overexpressing it increased the m6A modification level. However, the protein abundance of m6A methyltransferase METTL3 and demethylases FTO and ALKBH5 was unaffected. In addition, overexpression of miRNAs facilitates the binding of METTL3 on mRNAs, while miRNA downregulation impedes METTL3 binding to mRNAs. Taken together, these findings reveal that miRNAs could affect m6A methylation through modulating METTL3 binding to mRNAs.
Conclusions
To date, investigations about m6A have focused on the characteristic studies, including the discovery of its distribution profile and the “writer,” “eraser,” and “reader” proteins. One recent article revealed the crystal structures of the METTL3-METTL4 heterodimer and suggested that in the m6A MTase complex, METTL3 primarily functions as the catalytic core, while METTL14 serves as an RNA-binding platform (Wang et al., 2016); however, the studies regarding potential roles of m6A modifications in transcriptional regulation are still limited. In this context, there is a lot of information regarding m6A modification modulating ESC differentiation and SC reprogramming by regulating gene expression, while more is still needed to be investigated. The underlying mechanism is due to the regulation role of m6A modification in mRNA degradation, which further affects the gene expression. The m6A-mediated mechanism can provide a rapid posttranslational regulation manner to control gene expression at the RNA level.
According to the mapping data of m6A, numerous pluripotency and differentiation genes were observed having m6A modification, which suggests the potential role of m6A in cell fate determination (Geula et al., 2015). We assume that m6A RNA modification could regulate the cell fate by regulating the mRNA degradation, and the probable mechanisms can be summarized into three key points. First, during the cell development process, there is a competitive action between opposite lineage specifiers, whereafter the predominant lineage specification genes will determine cell fate (Montserrat et al., 2013; Shu et al., 2013).
Second, m6A modifications in the predominant lineage specification genes could regulate the expression of these genes and thereby affect the result of the cell fate competitive action. For instance, embryo-related specification genes are predominate in ESC at the naive state, and under this situation, the presence of m6A in embryo-related specification genes will promote degradation of the transcription of these genes and push ESC toward differentiation. Finally, altering the original m6A level could break the balance between different lineage specification genes and thus redetermine the cell fate. It means the downregulation of m6A level assists cells to maintain the previous state by promoting the expressions of the previous predominant lineage specification genes. Conversely, m6A upregulation decreases cell's previous characteristics, leading cells to develop in the opposite direction (Fig. 2).
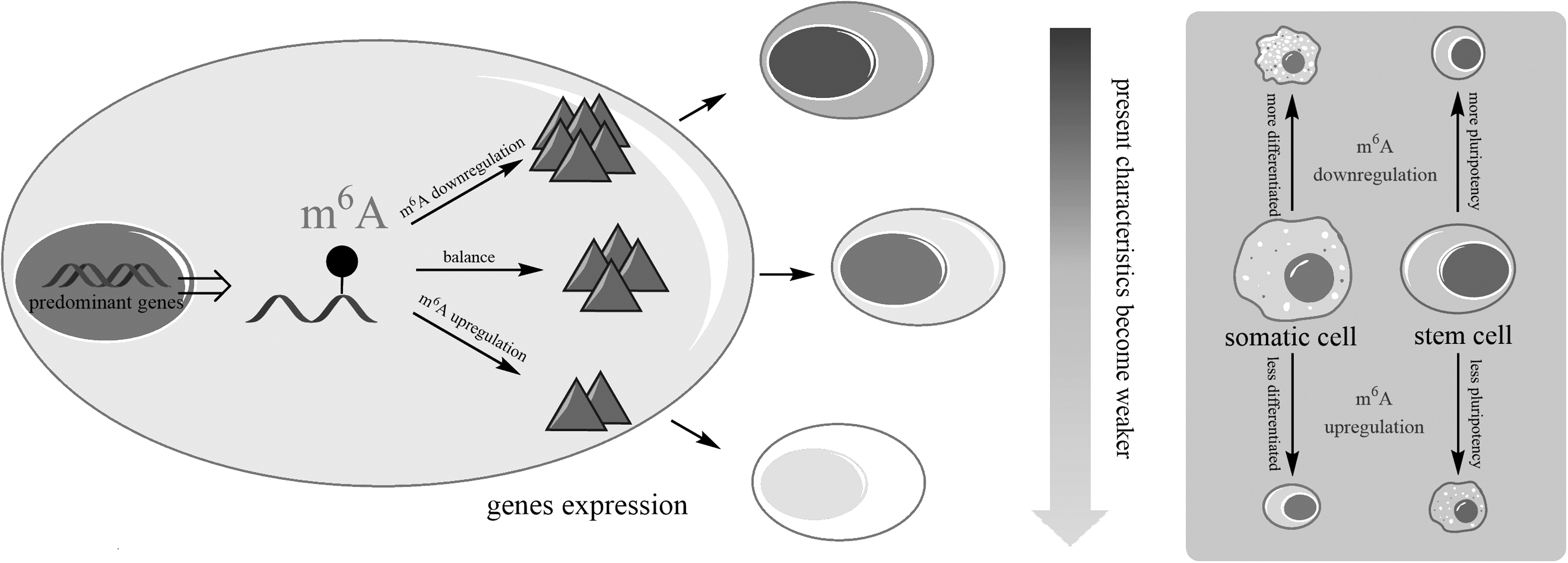
m6A level alters cell previous state by regulating predominant gene expression. m6A downregulation promotes predominant gene expression resulting in present characteristics enhanced, and its upregulation restrains predominant gene expression resulting in present characteristics weakened.
This hypothesis is concordant with the previous studies that decreased m6A could block ESC differentiation and increased m6A could promote MEFs reprogramming (Esteban et al., 2010). In addition, several recent studies show that m6A determines Drosophila sex via controlling female-specific alternative splicing of Sex-lethal (Sxl) by its reader protein YT521-B (Haussmann et al., 2016; Lence et al., 2016). This implies that m6A can not only determine cell fate by influencing the function of RNA degradation but also the RNA splicing, or even through other unfounded aspects. In conclusion, cell fate can be regulated by the alteration of m6A levels during the cell development. However, further work is still required to validate this hypothesis.
As MTases and demethylases regulate the alteration of m6A levels, most studies were focused on the former, while the latter, such as FTO and ALKBH5, is still limited. It would be significant to explore the cofactors of these demethylases, which influence their catalytic activity. It has been reported that L-ascorbic acid, also known as vitamin C, is a critical antioxidant and a cofactor for some Fe (II)- and α-ketoglutarate-dependent dioxygenases. Vitamin C could promote the generation of iPSCs through the mechanism of epigenetic change by modulating the activity of dioxygenases, including DNA hydroxylases (10–11 translocation, TET) and histone demethylases (Jhdm1a/1b) (Esteban et al., 2010).
As FTO and ALKBH5 are also Fe (II)- and α-ketoglutarate-dependent dioxygenases (Gerken et al., 2007; Thalhammer et al., 2011), we hypothesize that vitamin C could similarly play a critical role in the SC reprogramming by modulating the activity of these demethylases (Fig. 3). TET and Jhdm1a/1b related to DNA and histone protein modifications control gene expression by pretranscriptional regulation. Although posttranscriptional modifications are known to occur to RNAs, the impact of these modifications has only been explored recently (Yue et al., 2015). Also, it is attractive whether vitamin C could also affect posttranscriptional gene expression by impacting on the activity of FTO and ALKBH5 compared to pretranscriptional regulation. If so, it will provide a foundation for better understanding of the modulation mechanism of vitamin C in cell fate determination from epitranscriptional sites.
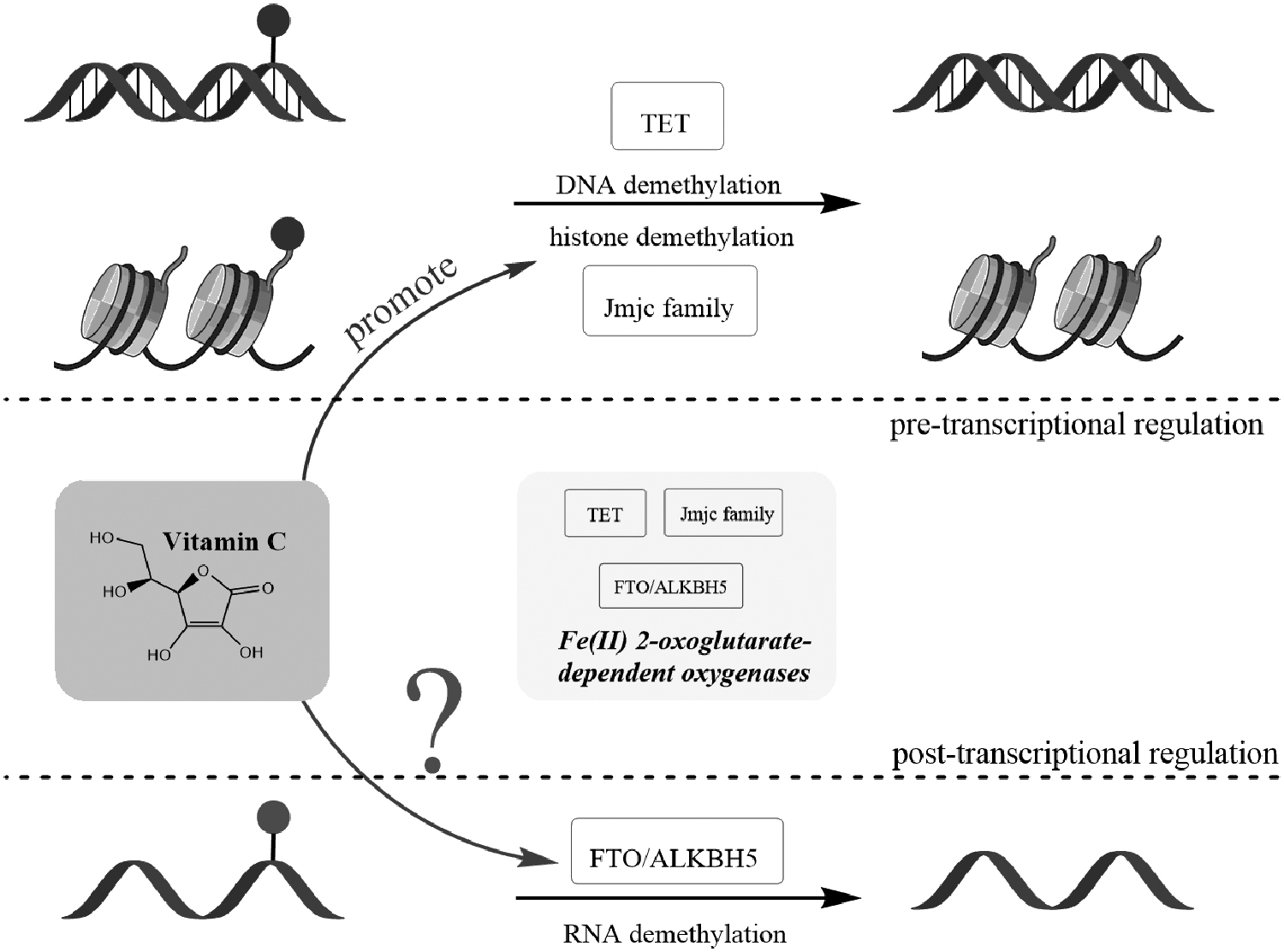
Vitamin C is a cofactor of some Fe (II)- and α-ketoglutarate-dependent dioxygenases, such as DNA and histone demethylases. Vitamin C regulates epigenetic reprogramming by modulating DNA hydroxylases (TET) and histone demethylases (Jhdm1a/1b), and we propose that it will also influence the function of RNA demethylases (FTO/ALKBH5), which belonged to Fe (II)- and α-ketoglutarate-dependent dioxygenases as well.
Footnotes
Acknowledgments
This work was supported by the Youth Fund of the National Natural Science Foundation of China (81400935) and the China Postdoctoral Science Foundation (2012M510302). We thank the support provided by the professors in the College of Life Sciences and Bioengineering, Beijing University of Technology, and especially thank Dr. Sakhawat Ali for his language help on the article.
Author Disclosure Statement
The authors declare that no conflicting financial interests exist.