
Abstract
Abstract
In this study, the distribution as well as the effect of autophagy on reprogramming in pig cloned embryos were observed immediately after somatic cell nuclear transfer. Results showed that the LC3 was at the highest level in cloned embryos at 2-cell stage, and it decreased with the development from 2-cell stage to blastocyst. Different to cloned embryos, the intensity of LC3 in parthenogenetic activation (PA) embryos was at the highest level at 4-cell stage. A markedly higher level of Bmp15, H1foo, and Dppa3 was shown in cloned embryos at 2-cell stage (p < 0.05 or p < 0.01), but a significantly lower level of LC3, Sox2, and eIF1A was observed at 4-cell stage (p < 0.05), compared with PA embryos. When the efficient interfering by the LC3 siRNA was performed on the cloned embryos (p < 0.01), not only the mRNA level of maternal Cyclin B, Bmp15, Gdf9, c-mos, H1foo, and Dppa3 was increased significantly (p < 0.05), but also the expression of Dnmt1 and Dnmt3b was obviously upregulated (p < 0.05). Although the expression of Sox2 and Oct4 is not changed, the expression of Stat3 decreased significantly (p < 0.05). Furthermore with the treatment of 200 nM rapamycin, the expression of eIF1A and Stat3 was significantly increased at 4-cell stage. In conclusion, the LC3-dependent autophagy mainly occurred in cloned embryos at 2-cell stage, but at 4-cell stage in PA embryos. In addition, the modulation of autophagy could affect genome activation by influencing the degradation of maternal mRNA and regulating the expression of DNA methyltransferase.
Introduction
M
However, the overall efficiency remains extremely low (<5% depending on the species (Schatten et al., 2005), only a few reconstructed embryos are capable of developing to blastocyst stage (Jaenisch et al., 2005) and even the cloned offspring are prone to the postnatal physical abnormalities, including respiratory distress, heart failures, circulatory abnormalities, immune dysfunctions, and brain diseases (Schatten et al., 2005). A possible explanation for the developmental incompetence of SCNT embryos is inadequate reprogramming of the nucleus transplanted into an enucleated oocyte, which involves the normal transcriptional reactivation of embryonic genes (Armstrong et al., 2006; Kumar et al., 2007; Liu et al., 2012).
Autophagy is a basic system that degrades cytoplasmic macromolecules (proteins, lipids, and nucleotides, etc.) and organelles (e.g., mitochondria and endoplasmic reticulum) to recycle cellular components for maintenance of cellular homeostasis (Mizushima and Komatsu, 2011) and embryonic development (Mizushima and Levine, 2010). Recently, autophagy has been shown to involve in regulating the process of somatic reprogramming that autophagy could modulate homeostasis of pluripotency-associated proteins such as Oct4, Sox2, and Nanog (Cho et al., 2014). In addition, for early embryos, autophagy could not only change the level of DNA demethylation, but also degrade the unnecessary proteins, organelles, and maternal suppressors accumulated in the oocytes during the transition from oocyte to the embryo (Shen et al., 2015; Tsukamoto et al., 2008b).
Finally, embryo quality and developmental ability could be improved by induction of LC3-dependent autophagy (Lee et al., 2016). In SCNT embryos, many genes are inappropriately programmed, which might be responsible for the embryonic disorders. So regulation of autophagy is a potential way to ameliorate this situation. However, the precise role of autophagy during SCNT especially in pig is not fully understood.
During early embryogenesis, many maternal mRNAs play an important role in normal embryonic development. For instance, the regulated synthesis and destruction of Cyclin B assumes the key role for early embryonic cell cycle regulation (Hormanseder et al., 2013); H1foo (Oocyte-specific linker histone H1) participated in the formation of an open chromatin structure of donor cells, which is helpful to restore pluripotency (Jullien et al., 2010); and maternal Dppa3 (developmental pluripotency-associated protein 3) can be involved in chromatin remodeling by protecting the imprinted genes from active demethylation (Bakhtari and Ross, 2014).
But to activate the embryonic genome, maternal mRNAs were required to undergo general decay after their dominating the early development of the embryos by translation. It has also been shown that the autophagy is a possible way to degrade maternal mRNAs (Shen et al., 2015; Xu et al., 2012). It is essential to explore whether autophagy that occurred in pig cloned embryos has the similar effect, because of more complicated interaction between nucleus and cytoplasm in cloned embryos.
As we know, maternal mRNA and proteins are obviously degraded at around 2-cell to 4-cell stage in the embryos, which is known as the period of oocyte-to-embryo transition (Alizadeh et al., 2005; Shen et al., 2015), and this period is a crucial step for the embryo development. It is reported that many embryonic genes were expressed at 4-cell stage in pig (Cui et al., 2005). Therefore, it is necessary to study the precise role of autophagy during this transition in SCNT pig. The objective of our study was to investigate the autophagic changes in the early development stage of porcine cloned embryos, and discuss whether the regulation of autophagy could affect the activation of embryonic genome and the degradation of maternal mRNA.
Materials and Methods
Chemicals
All chemicals were purchased from Sigma-Aldrich Co., Inc. (St. Louis, MO) unless otherwise indicated. All manipulations were performed on a heated stage adjusted to 38.5°C, except otherwise indicated. All procedures, including the collection of pig fetal fibroblasts from the 24th day (day 24) fetuses, were performed in accordance with animal welfare and ethics under the International guidelines for Biomedical Research Involving Animals.
Preparation of somatic cells for nuclear transfer
After thawing, the pig fetal fibroblasts established from day 24 fetus of Erhualian pig were cultured in Dulbecco's modified Eagle's medium (DMEM; Gibco BRL, Grand Island, NY) supplemented with 10% fetal calf serum (FCS; Gibco, Auckland, New Zealand) at 37°C in a humidified atmosphere containing 5% CO2.
After 1 week in vitro culturing, the confluent pig fetal fibroblasts were washed twice with Ca2+ and Mg2+-free PBS (Hyclone, Logan, UT), and those cells were further incubated at 37°C with 500 μL Trypsin-EDTA (0.5 g/L Trypsin and 0.2 g/L EDTA; Gibco BRL). Three minutes later, 500 μL DMEM supplemented with 10% FCS was added with pipetting. Subsequently, the medium containing the cells was transferred to an Eppendorf tube for centrifugation for 3 minutes (572 g). Then the supernatant was discarded and 500 μL HEPES-buffered TCM-199 supplemented with 2% cattle serum (CS; Gibco) (T2) was added for the fibroblasts suspension, which was stored at room temperature (22°C–24°C) until fusion.
Oocyte collection and in vitro maturation
Ovaries were obtained from prepubertal gilts at a local abattoir and were transported within 4 hours to the laboratory in 0.9% (w/v) NaCl at 37°C. In vitro oocyte maturation was performed as previously described (Li et al., 2009). Briefly, cumulus–oocyte complexes (COCs) were aspirated from 2 to 6 mm follicles using an 18-gauge needle connected to a 20-mL disposable syringe. COCs were selected according to their morphologic characteristics, that is, showing at least three layers of compact cumulus cells and evenly granulated ooplasm. After washing three times in TCM-199 plus 0.8 mM
Embryos production
After maturation, cumulus cells were removed by repeated pipetting in 1 mg/mL hyaluronidase. Then oocytes were placed in a 20 μL T2 drop prepared in the lid of a 60-mm diameter Petri dish (Corning, NY), which was covered with mineral oil.
For the production of parthenogenetic activation (PA) embryos, matured oocytes were washed twice in a F+ activation solution consisting of 0.3 M mannitol, 0.1 mM MgSO4, 0.05 mM CaCl2, and 0.01% (w/v) polyvinyl alcohol (PVA). Then they were electrically activated in a BTX microslide 0.5-mm fusion chamber (model 450; BTX, San Diego, CA) covered with 500 μL F+ by using a single direct current (DC) pulse of 0.86 kV/cm for 80 μs.
Different from traditional SCNT techniques, handmade cloning (HMC) is a simpler way to produce cloned embryos without micromanipulation (Kragh et al., 2005). For the production of HMC embryos, the modified HMC procedure was performed as previously described (Kragh et al., 2005; Li et al., 2009). Briefly, zonae pellucidae of matured oocytes were removed by pronase treatment (3.3 mg/mL, 2 minutes). For manual bisection, zona-free oocytes were lined up in drops of 20 μL T2 supplemented with 2.5 μg/mL cytochalasin B (20 oocytes per each drop). Bisection was performed by hand under stereomicroscopic control with a microblade (Homemade), and then the demi-oocytes (the selected putative cytoplasts) were transferred into T2 drops until fusion.
After bisection, a two-step fusion procedure was performed as follows. For the first fusion, half of the total quantity of prepared cytoplasts was transferred into one drop of T2. Then ∼200 somatic cells were placed into another T2 drop. After a short equilibration, cytoplasts were individually transferred to 1 mg/mL of phytohemagglutinin for 2 to 3 seconds, and then each cytoplast was attached to one single somatic cell, which was preequilibrated in the T2 drop. Subsequently, cytoplast–cell pairs were aligned to one wire of a BTX microslide 0.5-mm fusion chamber with the somatic cell farthest from the wire by using an alternating current (AC) of 0.06 kV/cm in the F0 fusion medium (0.3 M mannitol supplemented with 0.01% [w/v] PVA). Pairs were then fused with a single DC pulse of 2.0 kV/cm for 9 μs. After being removed carefully from the wire, pairs were further incubated in T2 to observe whether fusion occurred.
One hour later, fused pairs were selected and were transferred in the F+ activation medium. By using a 0.06 kV/cm AC, a pair and another cytoplast were aligned on the wire of the fusion chamber. The second fusion, including initiation of activation was performed by a single DC pulse of 0.86 kV/cm for 80 μs. Fusion was checked again after 15 minutes in the T2 drop.
After completion of the second fusion/activation, the reconstructed embryos were transferred to the chemical activation medium with 10 μg/mL cycloheximide and 5 μg/mL cytochalasin B in PZM-3 medium (Yoshioka et al., 2002), covered with mineral oil, and were incubated for 4 hours at 38.5°C in 5% CO2/20% O2 to complete the chemical activation. After that, embryos were washed twice in PZM-3 and cultured individually in well of the wells [WOWs, (Vajta et al., 2000)] in 400 μL of PZM-3 culture medium covered with 400 μL mineral oil for 6 days at 38.5°C in 5% CO2/20% O2 incubator (HERAcell 150i CO2 Incubator; Thermo Fisher Scientific).
Transfection of LC3 siRNA
For the transfection of LC3 siRNA, first the second fusion in HMC procedure was performed in the F0 fusion medium. Both negative control siRNA and an effective interfering fragment of LC3-specific siRNA were designed and commercially obtained (GenePharma, Shanghai, China), and their sequences are shown in Table 1. The working concentration of LC3 siRNA, 400 nM was achieved by mixing 20 μM RNA-free water dilution of siRNA with F+ activation medium.
After completion of the second fusion, all reconstructed embryos were evenly divided into three groups. Subsequently, with a modified BTX microslide 0.5-mm fusion chamber, reconstructed embryos were activated according to a same procedure as PA, but in F+ activation medium, containing no siRNA, 400 nM LC3 siRNA, or negative control siRNA, respectively, at 25°C. The negative control is embryos with addition of siRNA which has no homology with the target gene sequence. After incubation at 25°C for about 2 hours in T2 containing no siRNA, 400 nM LC3 siRNA, or negative control siRNA, respectively, the same procedure of chemical activation and in vitro culture were performed as mentioned above. Then 2-cell embryos were collected to undergo the real-time quantitative polymerase chain reaction (qRT-PCR).
Rapamycin treatment on cloned embryos
Rapamycin as a typical inducer of autophagy was used in the present study. The cloned embryos after chemical activation were immediately cultured in the PZM-3 medium containing 200 nM rapamycin for 48 hours. Then 4-cell embryos were collected to undergo qRT-PCR.
Assessment of gene expression level
The relative abundance of LC3 and several genes related to embryonic development and reprogramming, such as maternal genes (Cyclin B, Bmp15, Gdf9, c-mos, H1foo, and Dppa3), DNA methyltransferase (Dnmt1, Dnmt3b), pluripotency genes (Sox2, Oct4), transcription factors Stat3, and genome activation marker eIF1A were evaluated in 2-cell and 4-cell stage. Ten embryos per group at 2-cell or 4-cell stage were used to extract the total RNA with the Dynabeads mRNA DIRECT Kit (Invitrogen, Thermo Fisher Scientific, Vilnius, Lithuania) following the manufacturer's protocol. First-strand total cDNAs were synthesized with HiScript® Q RT SuperMix for qPCR (Vazyme Biotech Co., Ltd., Nanjing, China) according to the manufacturer's instructions. Then, these cDNAs were diluted 1:1 in DPEC water before analysis.
The real-time PCR was performed using the AceQ® qPCR SYBR® Green Master Mix (Vazyme) by a standard protocol in a reaction volume of 20 μL. The reaction mixture contained 10 μL AceQ qPCR SYBR Green Master Mix, 0.5 μL forward primer (10 μM), 0.5 μL reverse primer (10 μM), 2 μL each sample and 6.6 μL DPEC water. StepOnePlus Real-Time PCR System (Applied Biosystems, Life Technologies, Carlsbad, CA) was used for the qRT-PCR. The reaction conditions were 1 cycle at 95°C for 5 minutes, followed by 40 cycles of 95°C for 10 seconds, and 60°C for 30 seconds, and all reactions were run in triplicate with independent RNA samples. The fold-change in mRNA expression was determined with the reference gene 18 seconds by using the 2−ΔΔCt method. The primers used are shown in Table 2.
Immunofluorescent staining
Cloned embryos were collected at 1-cell (0 hours, immediately after the electric activation), 1-cell (4 hours, just finishing the chemical activation), 2-cell (28 hours), 4-cell (52 hours), and blastocyst (148 hours), whereas PA embryos were collected as control. The collected embryos were fixed by 4% paraformaldehyde for 30 minutes at room temperature. After being washed for 15 minutes in Ca2+ and Mg2+-free PBS with 1% BSA (PB1), the fixed samples were permeabilized at room temperature in 0.5% Triton X-100 for 1 hour. Subsequently, embryos were blocked in PB1 at room temperature for 1 hour. The embryos were then treated with LC3-I/II antibody (diluted 1:100; Cell Signaling Technology, Danvers, MA) at 4°C overnight, but embryos in negative control group were transferred into PB1 instead of the first antibody.
After washing with PB1 for 15 minutes, the samples were incubated with Alexa 488-conjugated anti-rabbit IgG (diluted 1:100; Goodbio Technology, Wuhan, Hubei, China) for1 hour. In addition, Hoechst 33342 (10 μg/mL) staining was performed for nuclear staining. After several washes, embryos were put in a small drop of glycerol dripped on the glass slide for mounting. Meanwhile, Lanolin was slightly smeared on the four corners of the cover glass. After that, the cover glass was put on the glycerol, followed by gently pressing until the embryos in the glycerol got properly thin. Then slides were observed under a fluorescence microscope (Zeiss LSM 700 META; Carl Zeiss, Jena, Germany). Each experiment was repeated three times and at least 10 embryos in each group were examined.
To analyze the fluorescence intensity, embryos in control and treatment group were mounted on the same glass slide. The same parameters were used to normalize across the replicates. By using ImageJ software (NIH), “Regions of interest (ROI)” were established covering the entire cytoplasm after images were converted to grayscale, and then the mean fluorescence intensity per unit area within ROI of immunofluorescence images was assessed. The final average intensities for the control and treated embryos were determined by the respective average values of all measurements.
Statistical analyses
For each treatment, at least three replicates were performed. Except data of fluorescence intensity were analyzed by using one-way ANOVA followed by the Duncan multiple range tests. All data were subjected to independent-sample t-tests and the results are shown as mean ± SEMs. p < 0.05 indicates the significance. All analyses were performed using SPSS software v.20 (SPSS, Inc., Chicago, IL).
Results
The distribution pattern of autophagy in pig embryos
First, the fluorescence of LC3 was observed in cloned and PA embryos as shown in Figure 1A and B. Although immediately after electric activation (1-cell stage, 0 hours), no LC3 signal was detected both in HMC and PA embryos. After chemical activation (1-cell stage, 4 hours), a compact pattern of LC3 signal was observed near the donor nucleus in cloned embryos, but a relatively weaker signal of LC3 was observed in PA embryos after chemical activation. Subsequently, the fluorescence intensity of LC3 was analyzed from 1-cell (4 hours) to 4-cell stage, both in cloned and PA embryos, and results showed that the intensity of LC3 reached the highest level at 2-cell stage in cloned embryos, but at 4-cell stage in PA embryos. Finally at blastocyst stage, a relatively limited LC3 signal could be checked in few cells of cloned embryos and the similar distribution of LC3 was observed in PA embryos, but with relatively strong LC3 signal in more blastocyst cells.
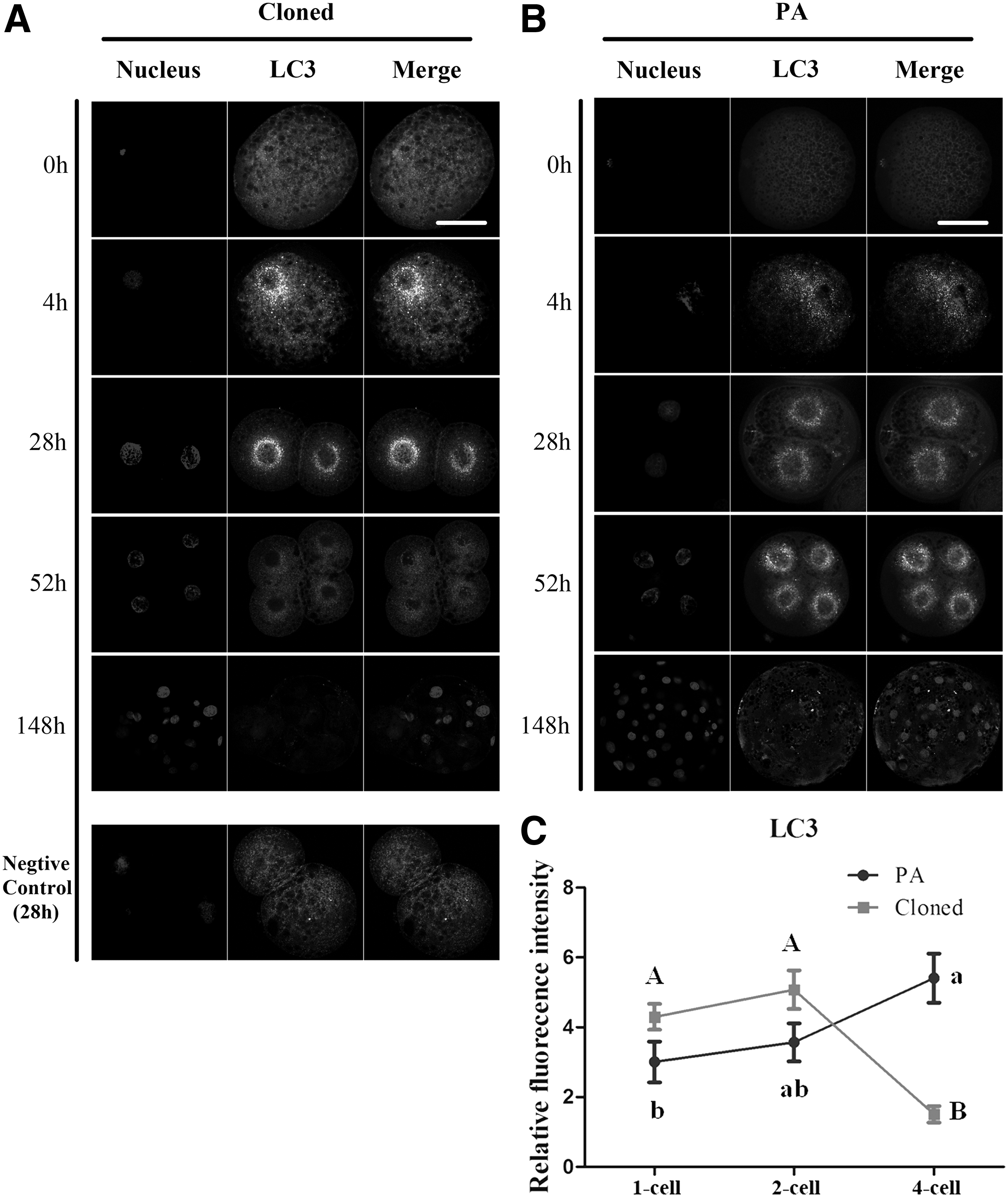
LC3-dependent autophagy in pig cloned and PA embryos.
Effect of LC3 siRNA transfection on cloned embryos
Although the other detected maternal genes and methylation- and pluripotency-related genes were at the similar level in cloned and PA embryos at the 2-cell stage, the expression of Bmp15, H1foo, and Dppa3 in HMC embryos was relatively higher than that in PA embryos (Cloned vs. PA: 1.2923 ± 0.0714 vs. 1.0024 ± 0.0485, 1.4319 ± 0.0865 vs. 1.0007 ± 0.0273, 1.2703 ± 0.0382 vs. 1.0120 ± 0.1066, p < 0.05 or p < 0.01, Fig. 2A). Furthermore, to evaluate the main function of LC3-dependent autophagy, the expression of LC3 was interfered as shown in Figure 2B. Results showed that LC3 could be significantly downregulated by all the three siRNA as shown in Table 1 (Control: 1.0035 ± 0.1022, Neg Control: 1.1695 ± 0.1861, sus-844: 0.4854 ± 0.0367, sus-1060: 0.4347 ± 0.1167, sus-948: 0.6759 ± 0.2257, p < 0.05 or p < 0.01).
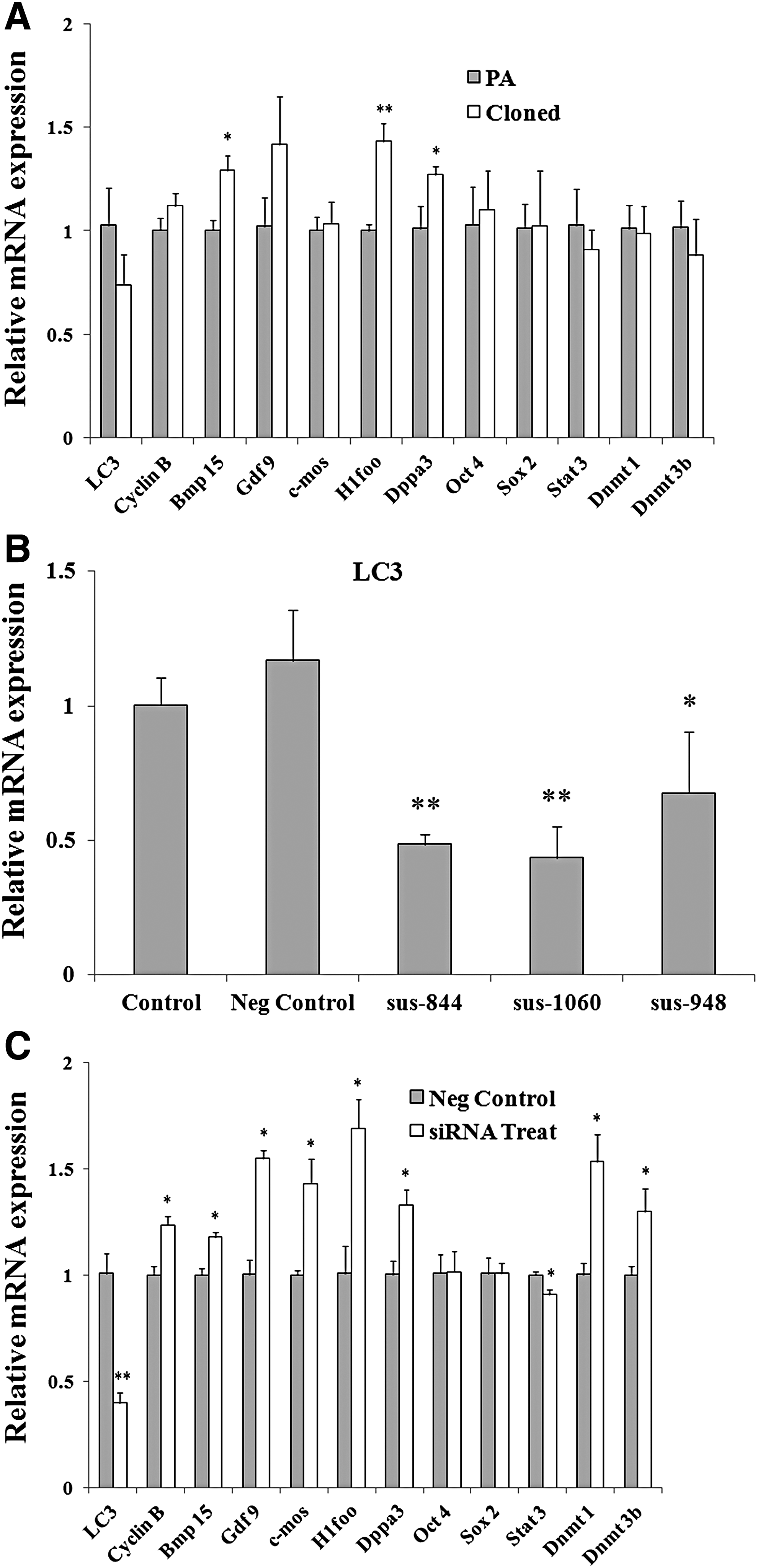
qRT-PCR analysis of several genes at 2-cell stage.
Subsequently, when the LC3 was downregulated by fragment “sus-1060” as shown in Figure 2C (Neg Control vs. siRNA Treat: 1.0089 ± 0.0927 vs. 0.3977 ± 0.0475), the relative mRNA expression of maternal gene Cyclin B, Bmp15, Gdf9, c-mos, H1foo, and Dppa3 and the expression of Dnmt1 and Dnmt3b were significantly increased at 2-cell stage (Neg Control vs. siRNA Treat: 1.0017 ± 0.0418 vs. 1.2369 ± 30.0427, 1.0005 ± 0.0321 vs. 1.1798 ± 0.0227, 1.0024 ± 0.0699 vs. 1.5503 ± 0.0386, 1.0004 ± 0.0201 vs. 1.4324 ± 0.1142, 1.0085 ± 0.1309 vs. 1.6909 ± 0.1379, 1.0042 ± 0.6387 vs. 1.3303 ± 0.0706, 1.0029 ± 0.0526 vs. 1.5347 ± 0.1287, 1.0022 ± 0.0379 vs. 1.2998 ± 0.1084, p < 0.05). On the contrary, the relative mRNA expression of transcription factor Stat3 was remarkably downregulated (Neg Control vs. siRNA Treat: 1.0004 ± 0.01636 vs. 0.9072 ± 0.0251, p < 0.05), whereas no change was observed on the expression of Sox2 and Oct4 with the LC3 interfering (Fig. 2C).
Effect of rapamycin on pig cloned embryos
With embryonic development from 2-cell to 4-cell stage, the expression of LC3, Sox2, and eIF1A in cloned embryos decreased significantly than that in PA embryos (Cloned vs. PA: 0.2932 ± 0.0521 vs. 1.024 ± 0.1590, 0.3681 ± 0.1281 vs. 1.0185 ± 0.1428, 0.6337 ± 0.0178 vs. 1.0028 ± 0.0523, p < 0.05, Fig. 3A). However, with the addition of 200 nM rapamycin, which could effectively induce autophagy (Sotthibundhu et al. 2016), the 4-cell cloned embryos in the treatment group showed a significant higher level of LC3 and eIF1A (Control vs. Rapamycin: 1.0261 ± 0.1708 vs. 2.5047 ± 0.2525, 1.0980 ± 0.0358 vs. 1.6689 ± 0.1989, p < 0.05 or p < 0.01). Although no difference of Sox2 and Oct4 was observed, the transcription factor Stat3 was also increased significantly with the treatment (Control vs. Rapamycin: 1.0127 ± 0.1169 vs. 1.0081 ± 0.0891, 1.0287 ± 0.1641 vs. 1.5848 ± 0.2084, 1.0036 ± 0.0844 vs. 1.6470 ± 0.0464, p < 0.05, Fig. 3B).
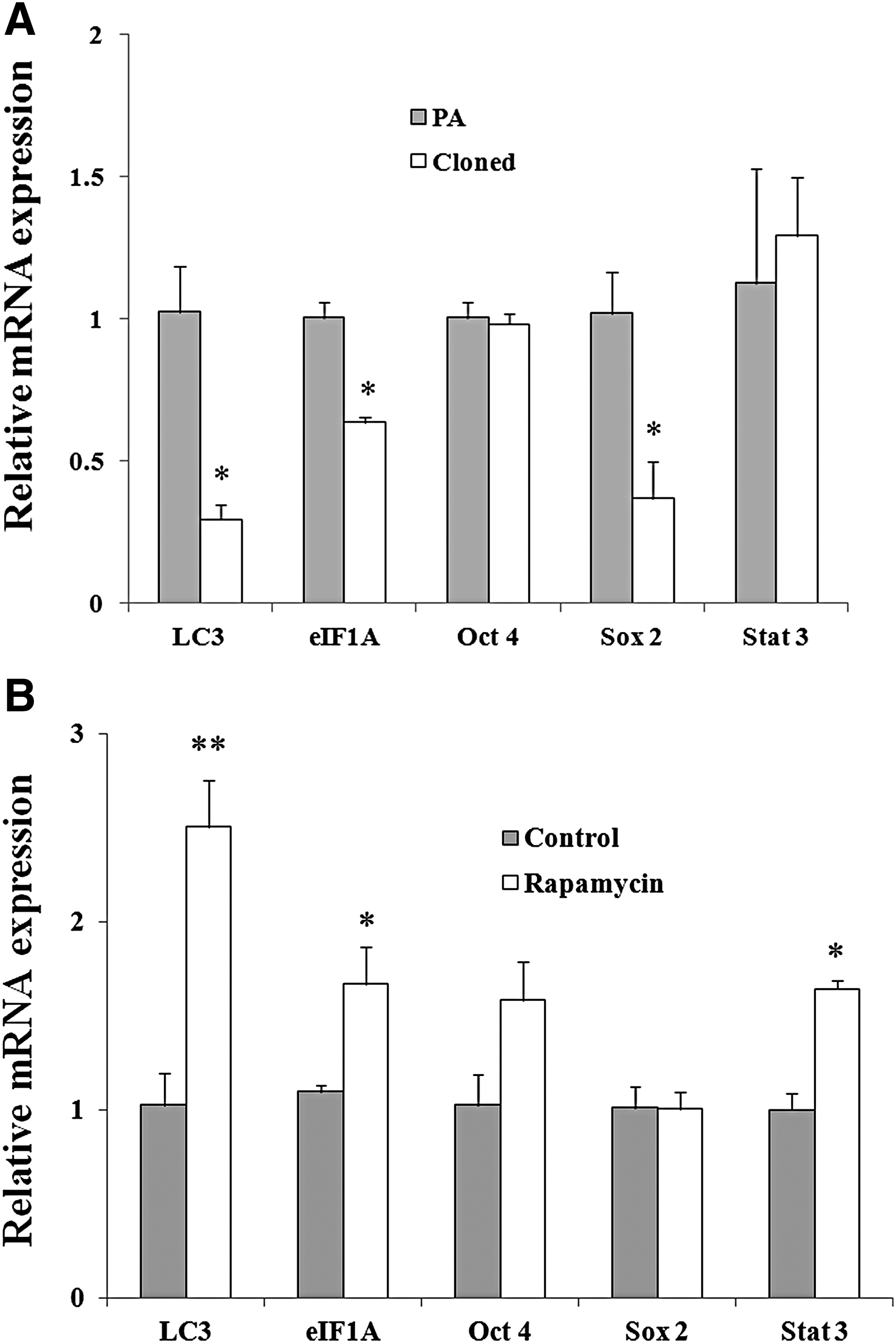
qRT-PCR analysis of genes at 4-cell stage
Discussion
The present study first confirmed that there is a significant decline in the level of LC3-dependent autophagy in cloned embryos from 2-cell to 4-cell stage. When the expression of genes in common cloned embryos compared with PA embryos, Bmp15, H1foo, and Dppa3 were higher at the 2-cell stage the LC3, Sox2, and eIF1A were lower at the 4-cell stage. However, once the LC3-dependent autophagy was interfering at 2-cell stage, the maternal Cyclin B, Bmp15, Gdf9, c-mos, H1foo, and Dppa3 were increased significantly. The expression of Dnmt1, Dnmt3b, and Stat3 was also obviously affected. However, when the LC3-dependent autophagy was induced at the 4-cell stage, the expression of eIF1A and Stat3 increased significantly. Therefore, the present study confirmed that the LC3-dependent autophagy occurred mainly at the 2-cell stage in pig cloned embryos, and it participates in the degradation of maternal mRNA and the regulation of epigenetic modification.
Several different reports about the distribution pattern of autophagy have indicated that LC3 seemed to be more evenly distributed in the cytoplasm of the cortex (Shen et al., 2015; Xu et al., 2012), but others showed that autophagy in somatic cells exhibits an intensive distribution around the nucleus (Liu et al., 2016; Wang et al., 2013), of which the similar pattern was observed in our study. When compared with PA embryos, the autophagy has a different timing of kinetics in cloned embryos. We hence think that this phenomenon may be caused by the different process of maternal genomic imprinting between parthenotes and cloned embryos (Jiang et al., 2007; Sepulveda-Rincon et al., 2016). Diploid parthenotes only possess maternal genetic information, and especially diploid parthenotes would not present the paternal imprinted genes.
In addition, SCNT embryos had difficulties in zygotic genome activation (ZGA) due to undefined epigenetic barriers preexisting in the genome of donor cells (Matoba et al., 2014). It is essential to break through the barriers of ZGA for reprogramming, which might be the reason for the topmost autophagy in pig cloned embryos at 2-cell stages before the ZGA.
After NT procedure, the donor chromatin needs to undergo epigenetic changes and modifications to get an embryonic-like chromatin structure (Martin et al., 2006; Pichugin et al., 2010; Yang et al., 2013a), all of which were governed by maternal factors that preexisted in the cytoplasm. The first major transcriptional event during embryogenesis occurred in 4-cell stage in pig (Magnani et al., 2008), and the expression of many genes changes from this period. So before 4-cell stage, elimination of a significant fraction of the mRNAs loaded into the egg by the mother should be carried out actively to trigger embryonic genome activation. Once the maternal mRNA cannot be degraded in time, many genes that are critical for early embryonic development cannot be expressed in time, resulting in the developmental retardation and female sterility (Yu et al., 2016b). Therefore, it is important to notice in the present that the LC3-dependent autophagy mainly occurred in cloned embryos at the 2-cell stage, a stage which is crucial for the degradation of maternal genes.
There are several different mechanisms to interpret the decay of maternal mRNA or ZGA. Some maternal-effect genes could indirectly or directly regulate this process (Yu et al., 2016a, b). The microRNA miR-430 could be also used as a potential link between ZGA and the decay of maternal mRNAs (Schier, 2007). Recently, the degradation of maternal proteins by the ubiquitin–proteasome pathway and autophagy has also been presented. As we know, autophagy is a crucial intracellular process that maintains cellular homeostasis (Tsukamoto et al., 2008a; Verlhac et al., 2010). So the regulation of autophagy will possibly affect other mechanisms to work on a compensation effect.
A few previous studies have shown that epigenetic modifications take place more rapidly in parthenotes (Adenot et al., 1997; Huang et al., 2016; Maalouf et al., 2008; Sepulveda-Rincon et al., 2016). There is an aberrant reprogramming of 5 mC and 5 hmC during early porcine embryonic development (Cao et al., 2014). Similarly in the current experiment, although the relative mRNA expression of Dnmt1 and Dnmt3b did not show a significant change in 2-cell cloned embryos compared with PA embryos, the level of H1foo and Dppa3 was significantly higher, and with the expression of LC3 decreased by siRNA, the expression of Dnmt1, Dnmt3b, H1foo, and Dppa3 was significantly upregulated at the 2-cell stage.
As we know, H1foo could quickly replace somatic histone H1 after transplantation, which will be of benefit to the formation of an open chromatin structure of donor cells. The expression of H1foo is stringently suppressed by DNA methylation in nonexpressing cells (Maeda et al., 2008); In addition, Dppa3 is one of the factors known to defend DNA methylation of the maternal genome and imprinted loci against active DNA demethylation processes (Nakamura et al., 2007). So, the relatively higher level of methylation-related genes in cloned embryos after the LC3 RNA interfering might indicate that autophagy also participate in the regulation of epigenetic modification.
Rapamycin is known to induce autophagy by inhibiting mTORC1 in mammalian cells (Kang et al., 2013; Thoreen et al., 2009; Yang et al., 2013b). With the elevated expression of LC3 by rapamycin at the 4-cell stage, the expression of Stat3 and eIF1A was increased significantly. To our knowledge, Stat3 is an important cytoplasmic transcription factor involved in cytokine signaling, the activation of which promotes transcriptional activation of genes with specific Stat3 recognition sites (Levy and Lee, 2002). In addition, eukaryotic elongation initiation factor 1A (eIF1A) has been used as a marker for detecting zygotic gene activation in some mammalian species, such as mouse, bovine, human, and porcine embryos (De Sousa et al., 1998; Lindeberg et al., 2004; Magnani et al., 2008). Therefore, the upregulated Stat3 and eIF1A might indicate that LC3-dependent autophagy is responsible for the transcription of embryonic genes.
Taken together, these results suggest that autophagy in pig cloned embryos could adjust embryonic genome activation by influencing maternal mRNA degradation and the expression of DNA methyltransferase at the 2-cell stage. From our results, not only autophagy could be used as a marker for reflecting the process of embryonic genome activation, it will also be possible to undertake different roles in the different development period of embryos. These results will provide a further reference for studies about the precise role of autophagy in early development of pig cloned embryos and more studies on subsequent embryo development need to be done.
Footnotes
Acknowledgments
The authors thank Baobao Chen and Wang Yao for technical assistance. This work was supported by the National Natural Science Foundation of China (grant number 31402076), the Funding of Central University in Nanjing Agricultural University (KJQN201521), and collaborative innovation program of Huaian (grant number HAC2014018).
Author Disclosure Statement
The authors declare that no conflicting financial interests exist.