
Abstract
Abstract
The human umbilical cord Wharton's Jelly- and the bone marrow- mesenchymal stem cells (WJ-MSCs and BM-MSCs, respectively) and the newly identified dental pulp pluripotent-like stem cells (DPPSCs) are new sources for stem cells with prospective use in cell regeneration and therapy. These cells are self-renewable, can be differentiated into several lineages, and can potentiate the immune responses. We hypothesized that three-dimensional (3D) culture conditions and directed differentiation using specific signaling regulators will enhance an efficient generation of mesoderm (MD) lineage independent from the origin or source of the stem cells. For a period of 3-days, cell aggregates were generated in a serum-free media containing ascorbic acid, retinoic acid, and keratinocyte growth factor; sonic hedgehog and bone morphogenic protein-4 signaling were inhibited using small molecules. In all cell types used, the biochemical and molecular analysis revealed a time course-dependent induction of the mesodermal, but not endodermal or ectodermal makers. In this study, we utilized a novel and efficient serum-free protocol to differentiate WJ-MSCs, BM-MSCs, and DPPSCs into MD-cells. Successful development of an efficient differentiation protocol can further be utilized and expanded on to obtain MD- derivative cell lineages.
Introduction
O
Furthermore, they have immune modulatory nature that makes them a good candidate for potential allogeneic therapeutic usages (Batsali et al., 2013; Kalaszczynska and Ferdyn, 2015; Weiss et al., 2008). Other advantages including large cell titer, ease manipulation, and no associated ethical concerns or genetic modulations.
The adult mesenchymal stem cells (MSCs) are nonhematopoietic, multipotent stem cells with the capacity to undergo self-renewal and multilineage differentiation (Pittenger et al., 1999). Initially, the rat bone marrow-MSCs (BM-MSCs) were first reported by Friedenstein and his colleagues (Friedenstein et al., 1966). Subsequently, they were characterized in several organisms, including humans (Mimeault et al., 2007). MSCs are fibroblast-like cells localized within the perivascular niche of organs and are implicated in the tissue turnover hemostasis (Mendes et al., 2012). BM-MSCs are the best-characterized adult MSCs. During embryogenesis, these cells arise from the intra-embryonic aorta-gonad mesonephros region, as a diverse population relative to their neighboring hematopoietic or endothelial progenitor cells (Migliaccio et al., 1986; Tavian et al., 1999; Wang et al., 2008).
At later developmental stages, MSCs go through a migration process and are stored in the bone marrow (Campagnoli et al., 2001). During the migration process, some cells are believed to be trapped and colonize the gelatinous material of the Wharton's jelly forming the WJ-MSCs (Batsali et al., 2013; Mendes et al., 2012). Thus, BM-MSCs and WJ-MSCs may have originated from a common ancestor; yet, our studies and others have shown that these cells have distinctive differences in the differentiation potential, transcriptomic and proteomic profiles (Ali et al., 2015; Fong et al., 2011; Hsieh et al., 2010).
Further, WJ-MSCs are contemplated to be more primitive and own features between embryonic and adult stem cells (Bongso et al., 2008). On the other hand, the dental pulp pluripotent-like stem cells (DPPSCs) have embryonic-like phenotypes with pluripotent characteristics. Unlike the dental MSCs, DPPSCs are small-sized cells with large nuclei and low cytoplasmic content (Atari et al., 2011, 2012b). Importantly, DPPSCs need restricted cell culture conditions and low cell density growth to maintain their pluripotent capacity, otherwise, at high cell density they show multipotent characteristics (Atari et al., 2012b; Nunez-Toldra et al., 2017).
Unlike the conventional 2D- cellular culture or differentiation protocols, three-dimensional (3D) conditions create a spatial environment, mimicking the in vivo cell niche, which enhances cell–cell contractions, extracellular matrix secretion, and the flow of nutrients between cells (Chen et al., 2006; Provin et al., 2008). This approach has been applied to PSCs including embryonic- and induced pluripotent stem cells, which have been successfully differentiated into mesoderm (MD) and its derived lineages following extensive in vitro induction protocols (Al Madhoun et al., 2011, 2013; Mummery et al., 2012; Voronova et al., 2013).
Using the 3D-approach, the mesodermal potential of WJ-MSCs, BM-MSCs, or DPPSCs has not been examined. To date, the differentiation protocols are variable due to differences in the molecular signature of these cells. The current protocols are mainly dependent on 2D-differentiation conditions; the use of prolonging serum-rich media that activate nontargeted biochemical signaling leading to poor differentiation (Martin-Rendon et al., 2008b) (Martin-Rendon et al., 2008a). On the other hand, an efficient and successful directional differentiation to the MD-lineage is a vital phase contributing to the efficiency of the downstream lineages, such as cardiomyocytes or hematopoietic cells.
In this article, we present a novel differentiation protocol to generate MD from WJ-MSCs, BM-MSCs, and DPPSCs. Independent from the stem cell source, the 3-days protocol achieved successful enrichment of cells expressing the MD- markers including the transcription factors Brachyury T (BraT), Mix Paired-Like Homeobox (MixL1) and Heart and Neural Crest Derivatives expressed 1 (Hand1), but not the endodermal or ectodermal markers. Thus, MSCs and DPPSCs could provide an excellent platform for MD- and downstream lineage differentiation, which highlights their potential role in clinical and basic science stem cell applications.
Materials and Methods
Ethical approval and procurement of human samples
The study was approved by the Ethical Review Committee at the Dasman Diabetes Institute (protocol No: RA-2013-009) in accordance with the World Medical Association Declaration of Helsinki- Ethical Principles for Medical Research Involving Human Subjects and Samples. Human umbilical cord matrix, Wharton's Jelly, WJ-MSCs and human BM-MSCs were purchased from ATCC (PCS-500-010 and PCS-500-012, respectively). DPPSCs samples were isolated from patients after signing informed consents at the Regenerative Medicine Research Institute, UIC Barcelona, Spain. All experiments were performed in accordance with the guidelines on human stem cell research issued by the Committee on Bioethics of the UIC Barcelona.
WJ-MSCs, BM-MSCs, and DPPSCs culture and maintenance
WJ-MSCs and BM-MSCs were cultured in DMEM/Hams's F-12 (1:1 vol/vol; Invitrogen) Medium complemented with 10% MSC-qualified (Invitrogen) fetal bovine serum (FBS), penicillin (100 U/mL; Life Sciences), and streptomycin (100 μg/mL; Life Sciences).
In this study, the used DPPSCs clones were previously isolated and characterized by our group as previously described (Atari et al., 2012b; Nunez-Toldra et al., 2017). In brief, immediately after extraction, the third molars were washed using gauze soaked in 70% ethanol and sterile distilled water. The molar pulp tissues were extracted from the dental pulp using a nerve-puller file 15 and forceps. Cells were isolated by digesting the pulp tissue with collagenase type I (3 mg/mL; Sigma) for 60 minutes at 37°C.
The cells were then separated using an insulin syringe and centrifuged for 10 minutes at 1800 rpm. The cell fraction was washed twice with 1 × phosphate-buffered saline (PBS). Once collected, the cells were counted and seeded in DPPSC medium. Primary cell lines were established and the medium was changed every 4 days.
During the splitting/passages of DPPSCs, cell density was maintained at 80–100 cells/cm2. DPPSCs were cultured in precoated flasks with 100 ng/mL fibronectin (Life Technologies) in a medium consisted of 60% DMEM-low glucose (Life Technologies) and 40% MCDB-201 (Sigma-Aldrich); supplemented with 1 × SITE Liquid Media Supplement (Sigma-Aldrich); 1 × linoleic acid-bovine serum albumin (Sigma-Aldrich); 10−4 M L-ascorbic acid 2-phosphate (Sigma-Aldrich); 1 × penicillin-streptomycin; 2% FBS (Sigma-Aldrich); 10 ng/mL hPDGF-BB (Abcam); 10 ng/mL EGF (Sigma-Aldrich); 1000 U/mL LIF (Millipore); chemically defined Lipid Concentrate (Life Technologies); 0.8 mg/mL BSA (Sigma-Aldrich); and 55 μM β-mercaptoethanol (Sigma-Aldrich).
Differentiation assay
All experiments were accomplished in triplicate. MSCs and DPPSCs (passages 2–4) were differentiated into mesoderm lineage following the protocol described for the human embryonic stem cells (Pagliuca et al., 2014) with major modifications to suit their developmental stage. At day 0, WJ-MSCs, BM-MSCs, or DPPSCs cultures (70%–80% confluent) were dissociated into single cells and resuspended in the differentiation media (MCDB131 media complemented with 8 mM D-(+)-Glucose, 14.6 mM NaHCO3, 1% fatty acid free-BSA, 2 mM Glutamax, 1% Pen/Strep, 1:200 ITS-X in PBS, 250 μM ascorbic acid, and 50 ng/mL KGF/FGF-7 (R&D Systems), 2 μM retinoic acid, 0.5 μM cyclopamine-KAAD (Calbiochem), 2 μM LDN193189 hydrochloride (Sigma), and 0.25 μM Phorbol 12,13-dibutyrate (Sigma).
For the generation of spheroidal colonies; cells (1.0–1.2 × 106) were added to a well of the 8-wells AggreWell Plate (Stem Cell Technologies; each well containing 1200 micro-wells, accordingly, each cell aggregate was generated from 800–1000 cells) (Stover and Schwartz, 2011a, 2011b) or aggregated in hanging drops cultures (800–1000 cells/20 μL drop)(Al Madhoun et al., 2013), and then incubated at 37°C with 5% CO2. At differentiation day 1, the produced cell aggregates were harvested, resuspended in a fresh differentiation media and transferred into Ultra-low adherence 6-well plates (Corning) at a lower density, about 300–400 aggregates/well. As shown in Figure 1, cells were harvested at differentiation days 0, 1, and 3 for RNA extraction or immunofluorescence analysis.
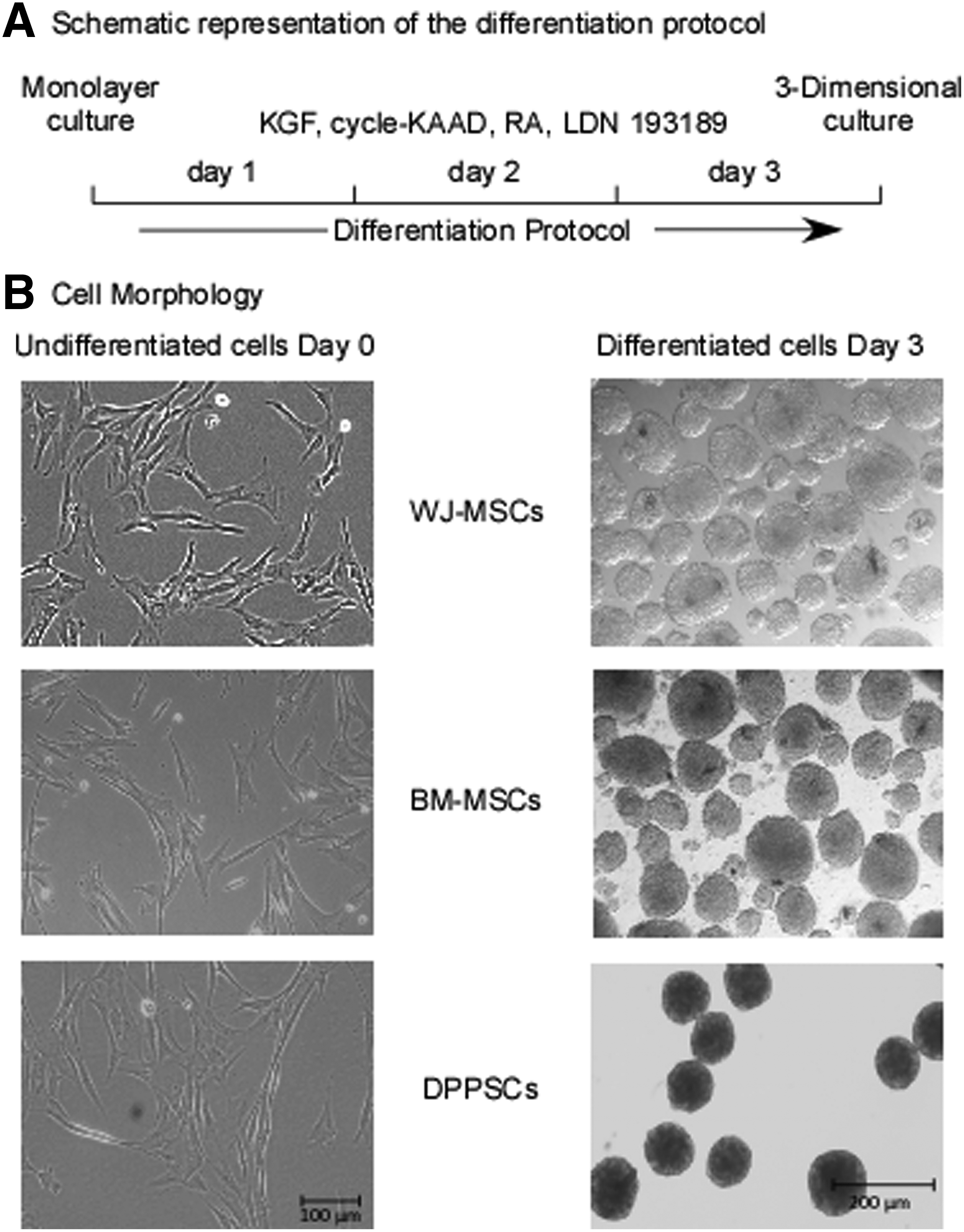
Adult and extraembryonic stem cells differentiation into MD-lineage.
Immunofluorescence assay
At day 3 of differentiation, cell aggregate sections were prepared and used for immunofluorescence assays using antibodies against BraT, MixL1, or Pax6. Cryosectioning was performed utilizing the protocol detailed (Al Madhoun et al., 2016; Gomes et al., 2010). At first, cells were rinsed with 1 × PBS and fixed in 10% formalin solution for 30 minutes at room temperature. Once this was done, cells were rehydrated using 1 × PBS for 15 minutes. Cells were then incubated in sequential series of sucrose solutions (10%, 20%, and 30%) for 30 minutes for each concentration. The sucrose solution was then removed and the cells were placed in a cryomould and covered with an OCT freezing gel.
The cryomould was subsequently frozen at −80°C. Once moulds were sufficiently frozen, OCT blocks were removed and cryosectioned using a cryostat (Bright OTF 5000 Cryostat, Hacker Instruments). Cryosections were mounted onto slides and heat dried to remove the OCT gel. For immunostaining, slides were washed with PBS and incubated overnight with the conjugated antibodies BraT, MixL1 and Pax6 antibodies (Table 1) were conjugated with Alexa Fluor- 488 or 594, respectively, using APEX Antibody Labeling Kits (Invitrogen) as described by the manufacturers.
BraT, Brachyury T; Hand1, heart and neural crest derivatives expressed 1; MixL1, mix paired-like homeobox.
RNA extraction, cDNA synthesis, and quantitative real-time polymerase chain reaction reactions
RNA purification kits (Norgen Biotek, Canada) were used for total RNA extractions following the manufacturer's protocol. QuantiTect Reverse Transcription Kit (Qiagen, Inc.) was used to prepare cDNA from 50–100 ng total RNA. Quantitative real-time polymerase chain reaction (qRT-PCR) assays were performed as previously described (Al Madhoun et al., 2011, 2013). Primer pairs with equivalent efficiencies are described in Table 2. Primer Bank was utilized to select the primers (Wang and Seed, 2003). qRT-PCR assays were performed using the ABI7900 system (Applied Biosystems) and SDS software. Comparative Ct was used to calculate relative gene expression, as previously described (Voronova et al., 2011, 2013). The results were normalized to the GAPDH- CT-values, and averages ± SEM are shown expressed relative to control or day 0 undifferentiated cells, as indicated.
Statistical analyses
All experiments and assays were done in triplicate. Results were compounded and statistical significance was estimated with a one-tailed Student's t-test assuming equal variance and error is standard error of the mean (*p < 0.05) (Al Madhoun et al., 2013).
Results
Our studies and others revealed that 3D cell culture enhances cell–cell contacts and mediates biochemical signal interactions between cells, a process that improves cellular reprogramming and differentiation (Al Madhoun et al., 2011, 2013, 2016). In addition, we previously reported that in monolayer cultures, MSCs have an elongated spindle-shaped fibroblast-like morphology (Ali et al., 2015). Similarly, at high confluency (>70%), DPPSCs display similar structural morphology and properties to that observed for MSCs (Atari et al., 2012a, 2012b; Nunez-Toldra et al., 2017) (Fig. 1). Remarkably, both MSCs and DPPSCs generated 3D spherical structures upon culturing in the differentiation media using the micro-AggreWells or handing drop techniques (Fig. 1).
To screen cells for MD directed differentiation, a time course of lineage-specific gene expression during 3 days' protocol was examined. qRT-PCR analyses revealed a significant induction of the mesodermal genes, but not the endodermal- or ectodermal genes, starting from day 1 and peaking at day 3 (Fig. 2). Relative to undifferentiated cells, at day 3, the expression levels of BraT transcripts were 38-, 44-, and 42- folds higher in the differentiated WJ-MSCs, BM-MSCs, and DPPSCs, respectively (Fig. 2). A similar trend was also observed for Hand1 and MixL1 gene expression in all stem cells studies. Contrary, the expression of the ectodermal genes, Sox1 and Pax6, in addition to the endodermal genes, GATA4 and Sox7, were either significantly reduced or did not show changes relative to day 0 expression (Fig. 2).
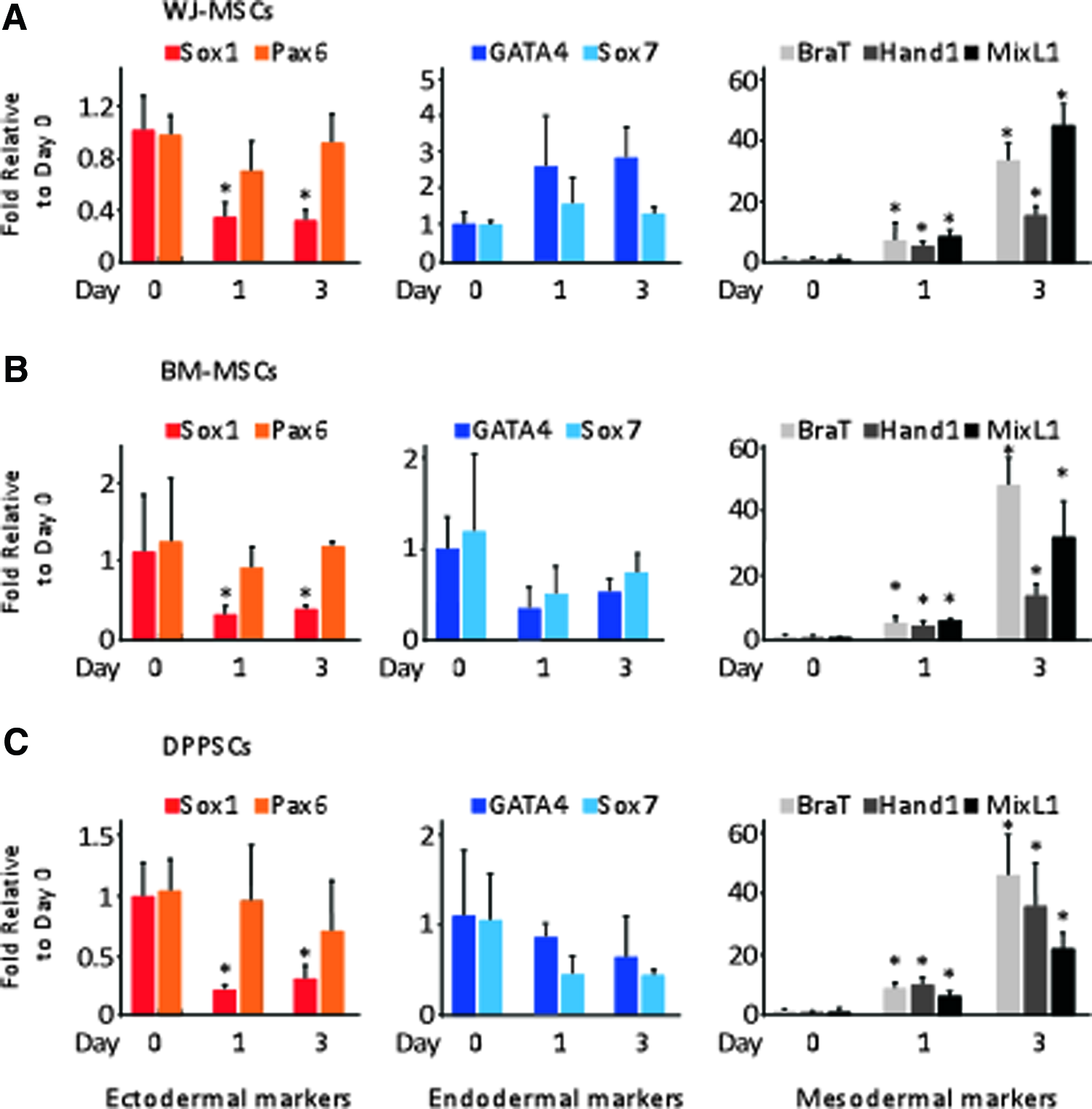
A time course differentiation protocol for
To validate the findings of the qRT-PCR analysis, we established the expression of the MD lineage-specific proteins via immunolocalization. Using fluorescent antibodies directed against BraT and MixL1, images detected the localization of proteins at the nuclei of day 3 differentiated WJ-MSCs, BM-MSCs, and DPPSCs (Fig. 3A, B). On the other hand, Pax6 specific antibodies failed to detect the protein in the cell clusters (Fig. 3C). Thus, our results suggest that the differentiation method led to a successfully enriched differentiation to MD lineage from all three different stem cell origins.
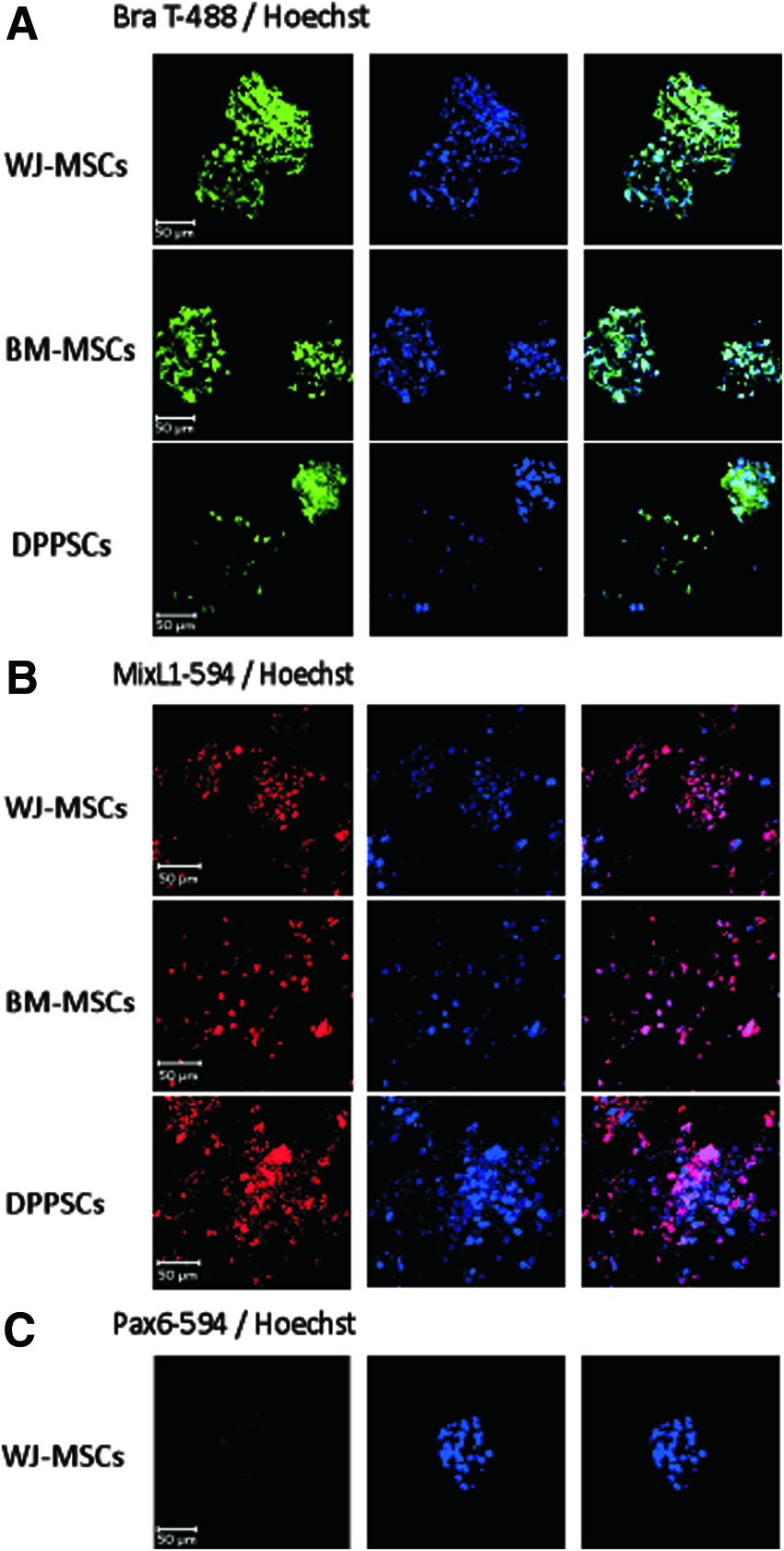
Immunofluorescence analysis for differentiated cells at day 3. Confocal laser images of immunofluorescence using
Discussions
The developmental plasticity of adult and extraembryonic stem cells to differentiate into multilineages has been previously described (Ali et al., 2015; Atari et al., 2012b; Deans and Moseley, 2000; Mennan et al., 2013; Ullah et al., 2015; Wang et al., 2004). On monolayer cultures, stem cells are poorly differentiated into the appreciated lineages, consequently due to the improper generation of MD; which is a crucial stage of the development of vital organs such as the heart or skeletal muscles (Al Madhoun et al., 2011, 2013). On the other hand, cell culture in 3D supports the generation of compact spherical structures, which enhance cell–cell interactions and mediate a network of biochemical and mechanical signals (Lin and Chang, 2008; Sart et al., 2014).
Under suitable conditions of directed differentiation, these cells can be directed toward specific lineage. This approach was previously reported for PSCs (Wong and Bernstein, 2010). In this report, we successfully differentiated stem cells, from different resources, into the MD-lineage within a short-timed protocol (3-days) by using a serum-free, chemically defined culture media and employing 3D-spherical body formation to enhance biochemical and mechanical interaction between cells.
The stem cells were cultured, for 3 days, in a differentiation media containing RA/KGF inducers and SHH/BMP signaling inhibitors, which are adequate to enrich the MD lineage from MSCs. This approach was also found to be applicable for DPPSCs obtained from a confluency culture. Studies have shown that KGF treatment mediates MSCs differentiation into the ectodermal derivative sweat gland-like cells (Xu et al., 2016) and improves the generation of mature T lymphocytes from thymocytes, MD progeny (Wils et al., 2012). RA mediates neurogenesis (Tonge and Andrews, 2010; Voronova et al., 2011) and hematopoiesis (Ronn et al., 2015) from stem cells when applied at high and low concentrations, respectively. Activation of SHH signaling pathway enhances osteogenesis and eliminates adipogenesis (Kim et al., 2010; Kondo et al., 2005).
BMP signaling was found to sustain stem cell self-renewal (Morikawa et al., 2016) and inhibit definitive endoderm induction in human ESCs (Vallier et al., 2009a, 2009b). On the other hand, the synergetic effect of these signaling pathways has a completely different effect relative to their mono-application. Kondo and his colleagues observed that SHH and high concentrations of RA promote the generation of sensory neurons (Kondo et al., 2005). During cardiac development, an active SHH signaling is not crucial to enhance cellular differentiation; nevertheless, a crosstalk with BMP4 is essential for a timely cardiomyogenesis.
In our protocol, the synergetic activation of RA and KGF signaling pathways enhanced MD formation. This differentiation process was further improved by the exit from self-renewal cycle through downregulation of BMP signaling and the inhibition of ectodermal/endodermal lineages by SHH suppression. Taking together, our differentiation protocol supports the notation that cellular niche is dynamic and dependent on the extracellular environment (Kumar et al., 2014; Xu et al., 2011).
Conclusions
In this study, we have developed a protocol to induce MD cells from three different types of stem cells at a high efficiency. We utilized a 3D environment and a serum-free media supplemented with a variety of growth factors and cytokines to trigger the extracellular signaling pathways sufficient for a directed differentiation. In addition to the known advantages of using adult and extraembryonic stem cells over other sources of stem cells, we report that WJ-MSC, BM-MSCs, and DPPSCs are greatly suitable for MD generation and prospective downstream lineage differentiation if cultured under optimal differentiation conditions.
Footnotes
Acknowledgments
This work was funded by the Kuwait Foundation for the Advancement of Sciences (KFAS) under project number RA-2013-009 for Ashraf Al Madhoun. The authors would like to thank Ms. Valerie Lopez Atizado for microtome and slide preparations. The authors would like Dr. Haitham Ghunaim and Ms. Yasmine Baradei for proof editing the article.
Author Disclosure Statement
The authors declare that no conflicting financial interests exist.