
Abstract
Abstract
Human-induced pluripotent stem cells (iPSCs) hold considerable promise for future biomedical applications. However, the generation, isolation, and establishment of an iPSC line still presents many challenges. In this study, we describe a simple yet highly efficient two-step method for the isolation, purification, and passaging of human iPSC lines that utilizes commercially available reagents. The first step adapts iPSCs to single cell culture and passage, promoting survival and self-renewal; the second step enables the isolation and purification of bona fide iPSCs from a mixed population using column-based positive selection of cells expressing pluripotency markers such as TRA-1-60. Using this method, we were able to purify iPSCs from cell preparations containing differentiated or unreprogrammed cells, and even to isolate iPSC lines directly from derivation plates. The iPSC lines generated by this method maintained their pluripotency and genomic stability, as demonstrated by trilineage differentiation and karyotype analysis. The method presented here could be adopted for high-throughput isolation and expansion of iPSC lines and facilitate the widespread use of iPSCs in future applications.
Introduction
T
Currently there exist two major bottlenecks in the process of iPSC generation. First and foremost, identification and isolation of bona fide iPSC colonies from derivation plates containing a mixture of cells dominated by unreprogrammed source cells and partially reprogrammed intermediates present a considerable obstacle and often requires training and expertise in advanced cell culture techniques. Current methods of identification include visual inspection of colony morphology, live alkaline phosphatase staining (Singh et al., 2012), or staining for pluripotent stem cell surface markers such as SSEA4 and TRA-1-60 (Asprer and Lakshmipathy, 2015).
Nevertheless, the manual isolation process remains highly subjective, and colonies being isolated are oftentimes contaminated with unreprogrammed or partially reprogrammed cells. To enable widespread applications requiring de novo iPSC derivation, more efficient methods for identifying, isolating, and purifying reprogrammed cells are needed.
The second bottleneck is the subsequent passaging and expansion of the isolated iPSCs to produce a useful and sustainable cell line. It has been suggested that newly generated iPSCs should undergo continued passaging in order for the cells to lose the episomes transfected during reprogramming (Okita et al., 2011), and for the cells to erase epigenetic memories from the source cell (Liang and Zhang, 2013). Currently, most laboratories employ mechanical dissection in the expansion and passaging of the new iPSC lines, which is labor intensive, time consuming, and requires technical skill and specialized equipment.
In this study we report a method that uses column-based magnetic beads for positive selection of iPSCs followed by enzymatic single cell passaging and expansion. This method aims to address both aforementioned issues by simplifying the isolation and passaging process, eliminating subjectivity in colony selection, and allowing many lines to be derived at once, thereby drastically reducing the time burden in iPSC generation.
Materials and Methods
iPSC generation through reprogramming of endothelial progenitor cells
iPSCs were generated from endothelial progenitor cells (EPCs) derived from blood mononuclear cells (MNCs) using the StemRNA-SR Reprogramming Kit from Stemgent (Cambridge, MA) following the manufacturer's protocol. Details for the derivation and reprogramming of EPCs have been reported elsewhere (Gao et al., 2017). iPSC colonies were cultured in NutriStem XF/FF medium (Stemgent) during reprogramming and the first two clump passages.
Adaption of iPSCs to single cell passage culture
Starting from passage 3, iPSCs were transitioned to Cellartis DEF-CS Culture System (Takara Bio USA, Mountain View, CA). This medium supports single cell passage and culture of iPSCs in a feeder-free and defined environment. For single cell dissociation, iPSCs were washed once with phosphate-buffered saline (PBS) without Ca2+ and Mg2+ (PBS–/–; Gibco, Gaithersburg, MD) and treated with TrypLE Select 1× (Gibco) for 5 minutes at 37°C followed with pipetting to ensure single cell dissociation.
The single cell suspension was then centrifuged at 200 g for 3 minutes, resuspended in DEF-CS medium, and seeded into six-well cell culture plates coated with COAT-1 (Takara Bio USA) at a density of 4.0–5.0 × 104 cells/cm2. Cells were maintained at 37°C, 5% CO2, and >90% humidity, and culture medium was changed every day, until the cells reached a confluence of 1.5–3.0 × 105 cells/cm2, which normally occurred 3–4 days postpassage. With each passage, cells were redissociated into single cells and transferred to a new tissue culture plate coated with COAT-1.
iPSC population purity assessment using flow cytometry
A total of 5 × 105 cells were incubated at room temperature for 30 minutes with the primary or isotype control antibodies in 200 μL PBS containing 2% fetal bovine serum (FBS). Stained cells were then washed three times with PBS containing 2% FBS, filtered through a 30 μm filter, and analyzed by a Guava easyCyte 8HT Flow Cytometer from EMD Millipore (Billerica, MA). The primary antibody used was SSEA4 monoclonal antibody (MC813-70) conjugated to Alexa Fluor 647 (Life Technologies, Carlsbad, CA). Data collection and analysis were performed using the InCyte program included in the guavaSoft software suite (ver. 3.1.1), and instructions from the manufacturer were followed.
iPSC purification by magnetic activated cell sorting
Anti-TRA-1-60 MicroBeads were obtained from Miltenyi Biotec (Auburn, CA). Separation was performed according to protocol provided by the manufacturer. In brief, cells in a mixture were dissociated into single cells (up to 2 × 106 total cells) and then magnetically labeled with microbeads. The labeled cell suspension was then loaded onto an MS column (Miltenyi Biotec). After washing three times with medium to remove unbound cells, the collected cells from positively selected fractions were then eluted from the column after being removed from the separator (magnetic field). The eluted cells were counted and plated for expansion. A schematic illustration of the magnetic activated cell sorting (MACS) separation process is shown in Figure 1.
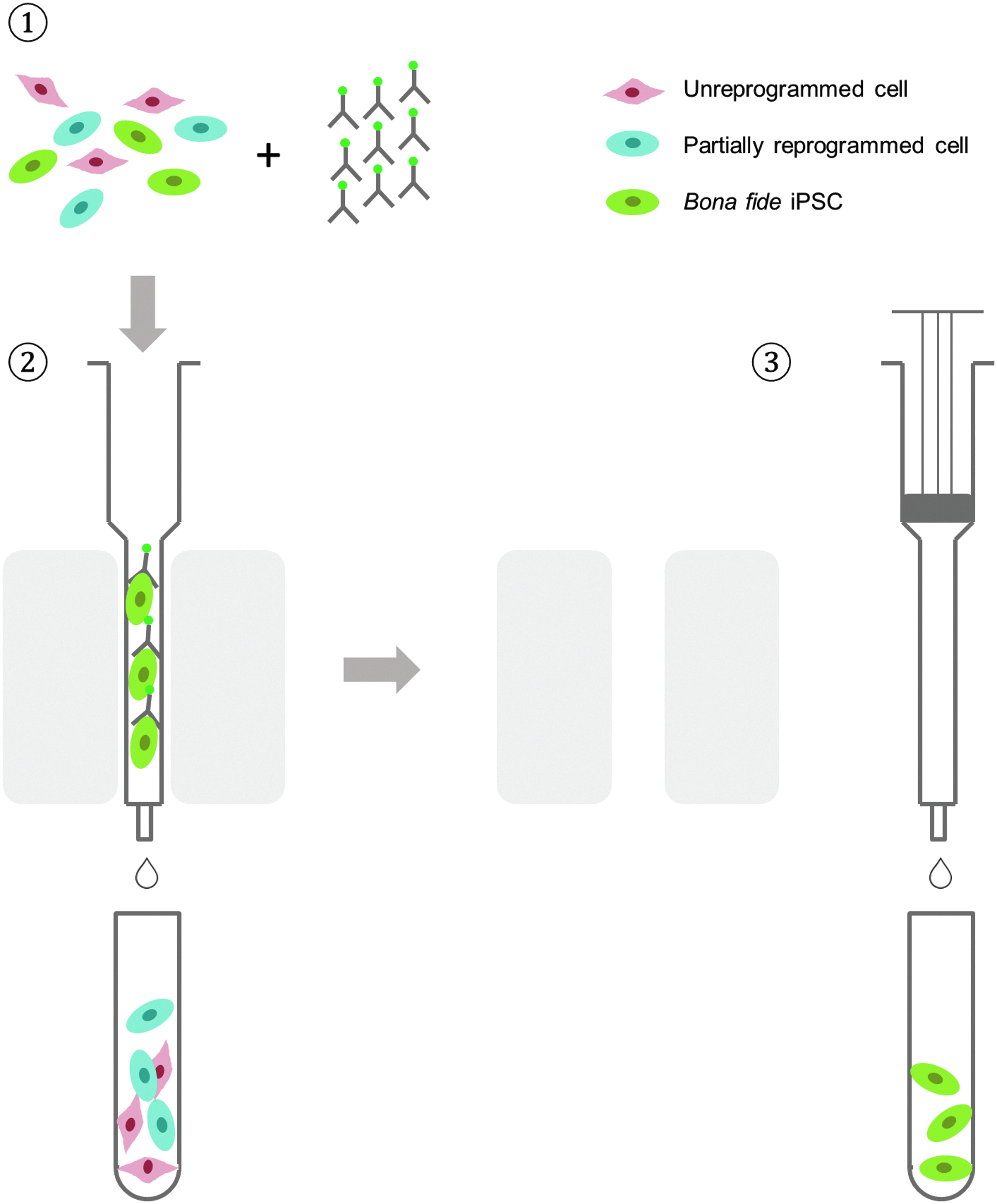
Schematic representation of iPSC purification by MACS. ➀ Cell mixture containing unreprogrammed somatic cells, partially reprogrammed cells, and completely reprogrammed cells is mixed with MACS MicroBeads coupled to antibodies targeted against iPSC surface makers (such as TRA-1-60). ➁ Cells are then loaded onto an MACS column placed in an MACS separator (a strong permanent magnet), and a high-gradient magnetic field is induced on the column matrix, which is strong enough to retain cells labeled with minimal amounts of MACS MicroBeads. Unlabeled cells pass through or are washed away from the column. ➂ Labeled cells are eluted from the column after removal of the column from the magnet. The entire procedure takes <30 minutes. iPSC, induced pluripotent stem cells; MACS, magnetic activated cell sorting.
Immunofluorescence staining
Cells were fixed using 4% v/v paraformaldehyde (Alfa Aesar, Tewksbury, MA), washed three times with PBS containing 0.2% v/v Tween 20 (PBST) (Fisher Scientific, Waltham, MA), and permeabilized using 0.15% v/v Triton X-100 (Sigma-Aldrich, St. Louis, MO) in PBS for 1 hour at 25°C. After permeabilization, cells were blocked with 1% v/v bovine serum albumin (Invitrogen, Carlsbad, CA) in PBST (PBSTB) for 30 minutes at 25°C. After gentle removal of PBSTB, cells were incubated with primary antibody in PBSTB overnight at 4°C.
Primary antibodies used in this study include OCT4 (Cell Signaling, Beverly, MA), SOX2 (R&D Systems, Minneapolis, MN), SSEA4 (Biolegend, San Diego, CA), TRA-1-60 (Biolegend), PAX6 (R&D Systems), SOX1 (R&D Systems), Brachyury (R&D Systems), α-SMA (Sigma-Aldrich), SOX17 (R&D Systems), and AFP (R&D Systems). After the overnight incubation, cells were washed three times with PBST and stained with secondary antibody (Alexa Fluor 488 or 594; Invitrogen) diluted 1:500 in PBSTB for 1 hour at 25°C. The cells were washed three times in PBST and stained with Hoechst dye (Invitrogen). Images of the stained cells were taken under a Leica DMi8 fluorescence microscope with a CCD camera.
Induction of differentiation
The iPSCs were subjected to suspension culture in embryoid body (EB) maintenance medium (iXCells Biotechnologies, San Diego, CA) to form EBs. The EBs were maintained in suspension culture for 1–2 weeks with daily medium change and attached to gelatin-coated dishes in the same medium for another 1–2 weeks before antibody staining.
Karyotype analysis
Cytogenetic analysis was performed on 20 G-banded metaphase cells per line by iXCells Biotechnologies.
Results
Generation of iPSCs by reprogramming EPCs using self-replicative RNA
EPCs derived from blood MNCs were reprogrammed using the StemRNA-SR Reprogramming Kit from Stemgent following a 30-day protocol (Gao et al., 2017). Colonies with characteristic iPSC morphology emerged about 20 days after self-replicative RNA transfection. Colonies were mechanically picked up between days 26 and 30 and were further passaged for expansion.
Purification of iPSCs
In the original reprogramming vessel, most iPSC colonies did not form clear edges and, therefore, were not well separated from the surrounding source EPCs (images not shown). Owing to technical difficulties in obtaining pure iPSC colonies by mechanical pickup, we observed that after the first passage (P1), iPSC colonies were contaminated by a small portion of unreprogrammed cells (Fig. 2A). The contamination was worsened upon further passaging. At the second passage (P2), more EPCs appeared in the culture than at the first passage and the cells lost their typical iPSC colonial morphology (Fig. 2B).
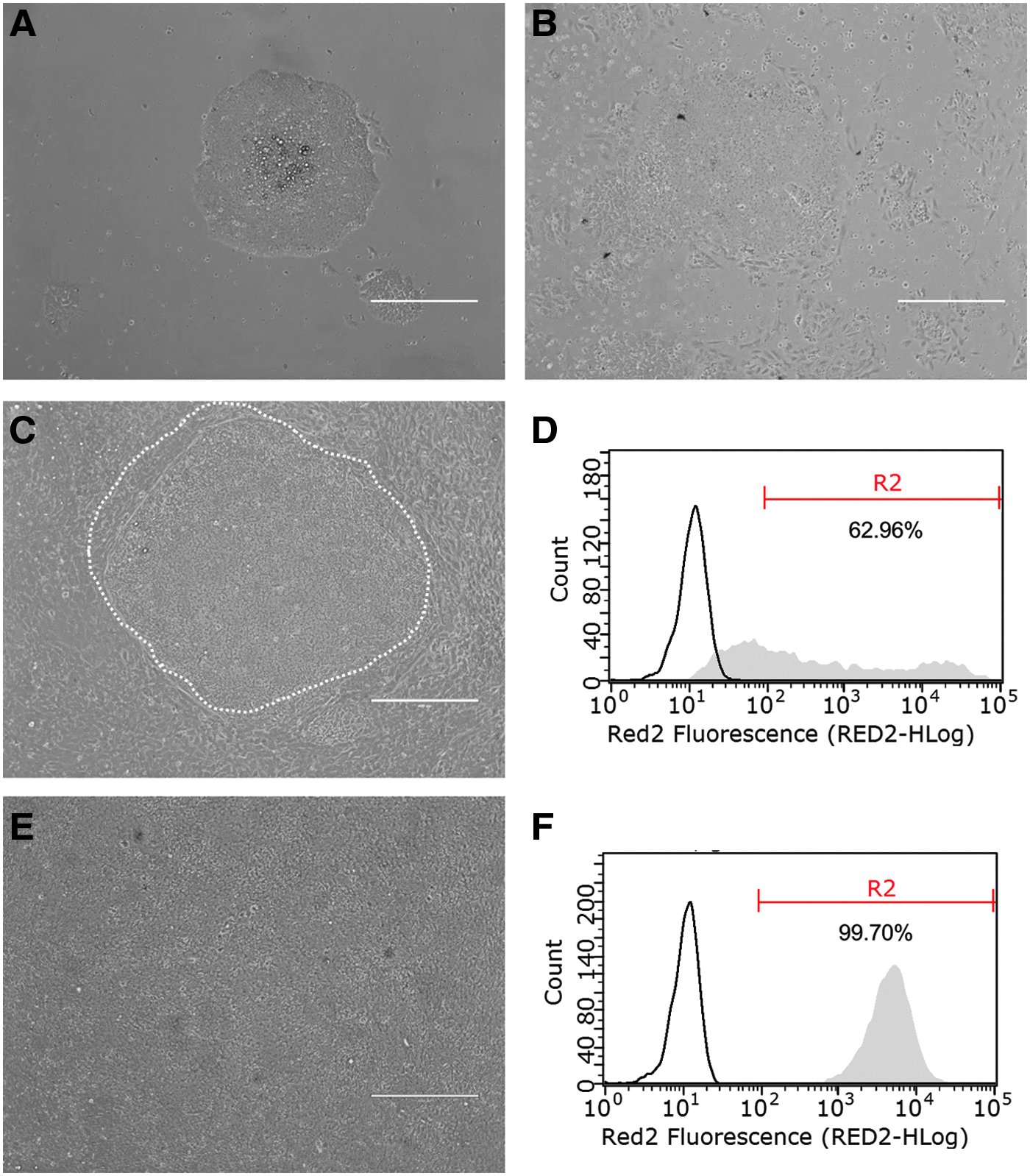
Purification of iPSCs by MACS.
To obtain a homogeneous and sustainable iPSC population, we chose to use MACS to purify iPSCs from the heterogeneous culture, which involves column-based positive selection of cells expressing pluripotency markers, for example, TRA-1-60 (Fig. 1). Purification by MACS requires iPSCs to be dissociated into a single cell suspension. In a preliminary study, it was found that dissociation of cells grown as colonies into single cell suspensions substantially reduced the cell viability to <40% (data not shown). Under the harsh condition during MACS purification, cell survival would be further compromised.
Therefore, to improve iPSC viability, we first transitioned the cells to a medium that supports single cell passage and culture, the Cellartis DEF-CS medium. Cells grown in the new medium (passage 3) typically show a patch of densely packed iPSCs surrounded by loosely packed somatic cells (Fig. 2C). Flow cytometric analysis of the cell population of six iPSC preparations (colonies picked from individual reprogramming experiments of six different source cells) showed the purity of iPSCs ranging from 5% to 75% (Table 1, L1 to L6). A representative flow cytometry histogram is shown in Figure 2D.
L1–L6 were purified at passage 2.
L7–L9 were purified directly from reprogramming plates.
iPSC, induced pluripotent stem cells; MACS, magnetic activated cell sorting.
MACS purification was accomplished with three simple steps (Fig. 1): labeling, loading and washing, and elution, which together took <30 minutes. After purification, the iPSCs were plated and single cell cultured until they reached the desired density (1.5–3.0 × 105 cells/cm2), usually within 4 days. Cells appeared as a homogeneous population with undifferentiated morphology (Fig. 2E). Flow cytometry analysis of the cell population revealed that all the six iPSC preparations had a purity of >95% (Fig. 2F and Table 1).
Direct isolation of iPSCs from reprogramming culture
iPSCs were also purified directly from the reprogramming plate. All cells in the well, including EPCs, iPSCs, and possibly partially reprogrammed cells, were dissociated from the well and applied onto the MACS column. Results of the purification of three iPSC lines in this way are also given in Table 1 (L7 to L9). Homogeneous iPSC populations were obtained in all cases.
Single cell passage and expansion
Once attained as a homogeneous population after purification, the iPSCs were further single cell passaged until passage 15. Cells expanded rapidly, usually reaching the desired density for further passaging in 3 days (Fig. 3A). Single cell passage and expansion retained pluripotency and genetic integrity of the iPSCs, as demonstrated in the next section.
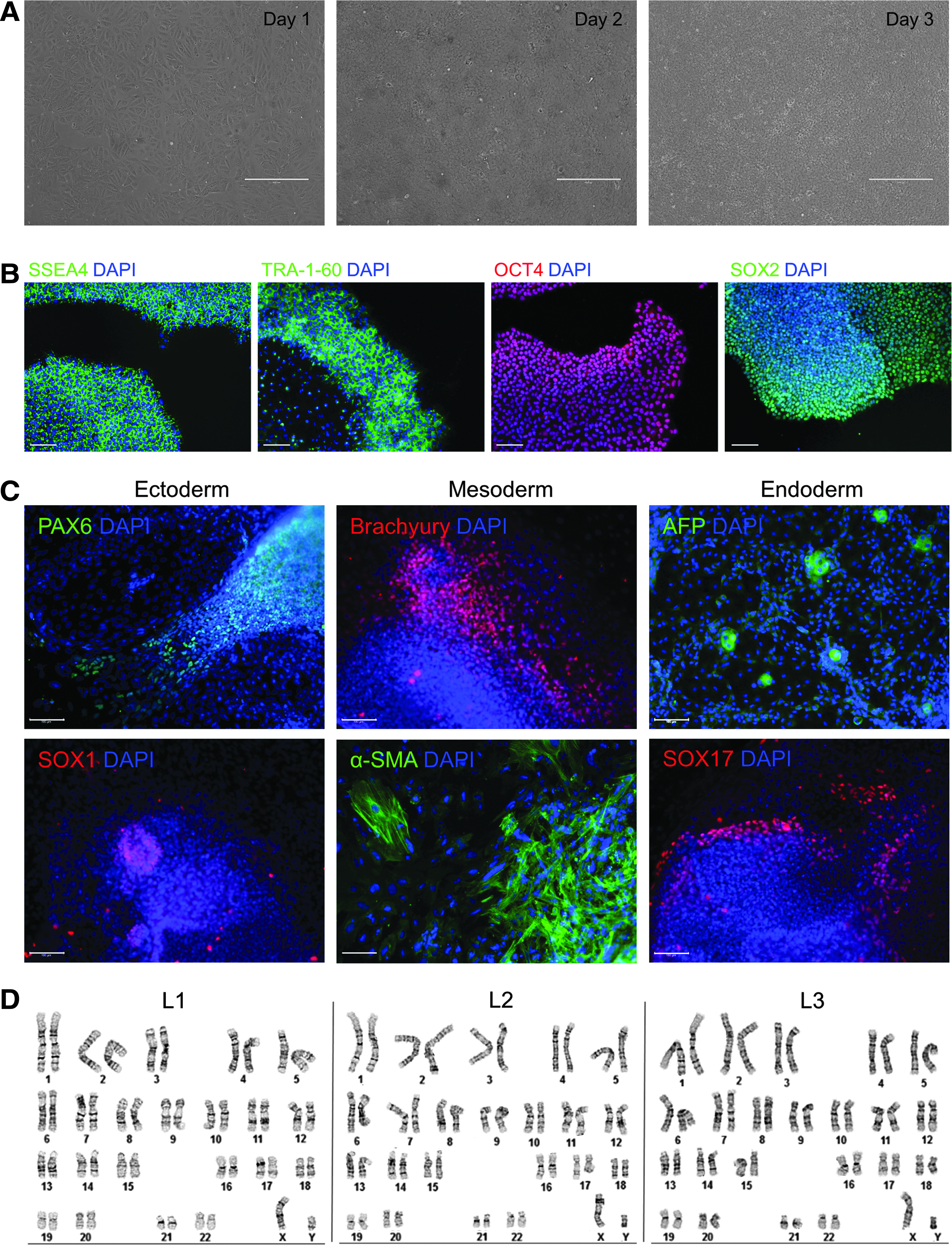
Single cell passage and characterization of iPSCs.
Characterization of the purified iPSCs
We characterized the MACS-purified iPSCs at passage 15. The cells expressed pluripotency markers SSEA-4, TRA-1-60, OCT4, and SOX2 (Fig. 3B). Also, these cells could form the three germ layers in vitro after EB formation, as demonstrated by the expression of ectoderm markers PAX6 and SOX1, mesoderm markers Brachyury and α-SMA, and endoderm markers AFP and SOX17 (Fig. 3C). Moreover, the cells maintained genomic stability after prolonged passaging in single cell culture (Fig. 3D).
Discussion
There has been a recent trend toward using magnetic beads that selectively bind to certain cell types for the isolation of specific cell populations. The so-called MACS was first described more than two decades ago (Miltenyi et al., 1990), and since then has been extensively used to isolate a wide variety of cell types (Grutzkau and Radbruch, 2010). More recently, it has been integrated into improved protocols for automated iPSC selection and passaging (Dick et al., 2011; Valamehr et al., 2012; Yang et al., 2015). Nevertheless, the application of MACS in iPSCs is still limited, and more studies are needed to demonstrate its usefulness in this burgeoning field.
In this study, we show that MACS can be used to purify iPSCs from cell preparations heavily “contaminated” by differentiated or unreprogrammed cells, and even to isolate iPSC lines directly from derivation plates, where iPSCs accounted for <10% of the total cell population. As such, the time burden of manually identifying and mechanically picking iPSC colonies is reduced drastically.
A prerequisite and key component of the method used here is single cell culture and passage of iPSCs. Typically, iPSCs are grown as colonies and passaged as clumps, as cell–cell contact is essential for cell survival and growth (Li et al., 2012). However, isolation by MACS requires a homogeneous single cell suspension. It has been observed that dissociation into single cells from a culture grown in conventional medium that supports colony growth substantially reduced cell viability, even in the presence of Rho-associated protein kinase (ROCK) inhibitors (data not shown), possibly because of losing survival and growth signals after single cell dissociation.
The Cellartis DEF-CS medium, recognized for its suitability for genome engineering (Valton et al., 2014) and single cell cloning (Feng et al., 2014), promotes reliable growth of iPSCs in a feeder-free and defined environment. Cells are grown in this medium as a homogeneous monolayer and are enzymatically passaged as single cells that maintain pluripotency with a stable karyotype (Asplund et al., 2016). We have found that iPSCs being transitioned to DEF-CS medium before MACS purification retained high viability (>80%).
A significant improvement of the method described here over the previously published methods (Dick et al., 2011; Valamehr et al., 2012; Yang et al., 2015) is the higher purification efficiency. In our method, purity of 90% or higher was achieved with a single round of MACS purification (Table 1), whereas in the method of Yang et al. (2015), lower purity (70%–80%) was obtained even after two to three rounds of MACS purification. The reason for this difference may lie in the fact that, in the previous methods, cells were grown in traditional ESC medium that supports colony formation.
When the cells were dissociated into single cells during MACS purification, these cells tend to form small clusters by nature and, therefore, some surface marker negative cells might be retained by the column if they clustered with marker positive cells. As such, the overall purification efficiency decreased. In contrast, in the method described here, cells were adapted to a medium that supports single cell culture and passaging before MACS purification. Cells remained as “true” single cell suspension throughout the purification process; therefore, only marker positive cells were retained by the column, hence the higher purification rate.
A critical technical detail for the success of the method is that after MACS purification and during single cell culture, the iPSCs should be plated at high density (4.0–5.0 × 104 cells/cm2) to maintain cell–cell contact to maintain genomic stability. It has been shown that iPSCs are susceptible to genomic abnormalities (Mayshar et al., 2010), especially during enzymatic single cell passaging (Bai et al., 2015). Maintaining cell–cell contact during passaging improves cell survival (Li et al., 2012), thus reduces cell stress and maintains genetic stability of the iPSCs.
It should be noted that direct isolation from the reprogramming culture will produce a polyclonal population of iPSCs, as individual clones are not maintained independently but are instead pooled together. Therefore, this method may have the drawback that individual clones cannot be analyzed or compared with one another. In contrast, the polyclonal system has the advantages of simplifying the derivation process, averaging all clones, and making the workload more manageable. If individual clones are desired, they can be derived by single cell cloning (Feng et al., 2014) from the polyclonal population.
Undifferentiated pluripotent stem cells express a number of surface antigens such as TRA-1-60 and TRA-1-81, which are downregulated upon differentiation (Andrews et al., 1984). It is possible to substitute the TRA-1-60 antibody (conjugated to the magnetic beads), used in this study, with any antibody directed toward a cell surface epitope specific to iPSCs, for example, TRA-1-81 and SSEA4, thus providing flexibility to the system. In addition, the system can be further modified to include a second column, with either positive or negative selection, to ensure almost complete removal of contaminating non-iPSCs. The combined use of both positive and negative selection has been successfully used previously for the detection and purification of fully reprogrammed iPSC colonies (Kahler et al., 2013; Quintanilla et al., 2014).
In this study, however, we showed that in some cases even though iPSC purity immediately after MACS may be <90%, the purity improved to >99% after continuous passaging in the Cellartis DEF-CS medium. This could be attributed to the fact that the Cellartis DEF-CS medium favors iPSC growth over other cell types; therefore, iPSCs eventually outgrow other cell types and become dominant in the population. Therefore, combined with the use of a growth medium that favors iPSC growth, a second column is not necessary to achieve a pure iPSC culture, given sufficient number of passages after MACS purification.
In conclusion, the results presented here suggest that MACS, combined with single cell culture and passage, has the potential to become an important technological development to support fully automated high-throughput iPSC derivation and culture methods.
Footnotes
Acknowledgments
We thank Dr. Marianna D. Solomotis for critically reading the article. This study was supported by internal funds of the U.S. Food and Drug Administration.
Disclaimer
The findings and conclusions presented in this article are those of the authors and do not necessarily represent views, opinions, or policies of the U.S. Food and Drug Administration.
Author Disclosure Statement
The authors declare that no conflicting financial interests exist.