
Abstract
Induced pluripotent stem cell (iPSC) technology refers to the reprogramming of terminally differentiated somatic cells into pluripotent stem cells by introducing specific transcription factors that are known to regulate pluripotency, including Oct4, Sox2, Klf4, and c-Myc. In this study, we reprogrammed the primary fibroblasts isolated from the Daxxflox/flox mice, which carry the Oct4-green fluorescent protein reporter, and employed wild-type littermates as a control to induce iPSCs, then knocked out Daxx by infecting with Cre virus at the cellular level. The pluripotency and self-renewal capacity of iPSCs were determined. In addition, Daxx deletion altered the pluripotency marker (Nanog, Oct4) expression and displayed neural differentiation defects. Particularly, by performing transcriptome analysis, we observed that numerous ribosome biogenesis-related genes were altered, and quantitative polymerase chain reaction revealed that the expression of rDNA-related genes, 47S and 18S, was elevated after Daxx deletion. Finally, we illustrated that the expression of the neurodevelopment-related gene was upregulated both in iPSCs and differentiated neurospheres. Taken together, we demonstrated that Daxx knockout promotes the expression of rDNA, pluripotency, and neurodevelopment genes, which may improve the differentiation abilities of mouse iPSCs (miPSCs).
Introduction
Differentiated mature somatic cells are reprogrammed to a multifunctional stem cell by transducing four specific exogenous transcription factors Oct3/4, Sox2, c-Myc, and Klf4, which all have characteristics similar to embryonic stem cells, called induced pluripotent stem cells (iPSCs) (Takahashi and Yamanaka, 2006). At present, the numerous studies related to iPSCs have mainly focused on transcriptional and epigenetic regulations (De Carvalho et al., 2010; Maherali et al., 2007). Spatiotemporal organization of the genome has been considered as an additional regulatory layer of chromatin, which is important for gene regulation and transcriptional ability (Gonzalez-Sandoval et al., 2013; Splinter and de Laat, 2011).
As a transcriptional regulator, the function of death domain associated protein (Daxx) on pluripotency maintenance and differentiation in miPSCs has not been addressed. Therefore, we studied the regulation of Daxx as a key molecule of chromatin regulation in miPSCs.
Daxx is a highly conserved, naturally disordered protein (Escobar-Cabrera et al., 2010) and widely exists in mammalian and human normal cells and tumor cells (Mahmud and Liao, 2019). The mouse Daxx protein consists of 729 amino acids that can be localized in the nucleus, PML-NB, heterochromatin, or cytoplasm (Bogolyubova et al., 2019; Li et al., 2000). Studies have shown that Daxx plays an important role in both antiapoptosis and proapoptosis as well as in DNA damage and repair (Meinecke et al., 2007; Michaelson et al., 1999; Shi et al., 2019; Yang et al., 1997; Yun et al., 2011; Zhang et al., 2010).
As a transcriptional regulatory protein in the nucleus, Daxx regulates transcription by binding to transcription factors, epigenetic modifiers, and chromatin remodeling factor (Best et al., 2002; Mahmud and Liao, 2019). Alternatively, Daxx is a molecular chaperone of histone H3.3; it localizes in the heterochromatin telomere region and assists in histone folding and transporting (Fang et al., 2018; Lewis et al., 2010; Voon et al., 2016). Despite the numerous studies regarding Daxx, its function is still unknown in pluripotent stem cells.
In this study, we established Daxx knockout (KO) iPSCs to research the function of Daxx on pluripotency, ribosome DNA, and neural development. In addition, we evaluated the expression of Daxx in the maintenance and differentiation capacity of iPSCs. Our results indicated that Daxx is essential for ribosome DNA transcription and neural development. Especially, this is the first description of the effect of knocking out Daxx on the characteristics and differentiation of iPSCs.
Materials and Methods
Animal and mouse embryonic fibroblast derivation
Male mice that contained the Daxx floxed allele were bred with female mice that expressed the enhanced green fluorescent protein (eGFP) under the control of the Oct4 promoter (Daxxflox/flox:Oct4-GFP) and were gifted by Prof. Fan HY (University of Zhejiang, Hangzhou, China). Heterozygous mice born in the same litter were used as a control group (Daxxflox/+:Oct4-GFP). Mouse genotypes were determined by polymerase chain reaction (PCR). All primary mouse embryonic fibroblasts (MEFs) were derived from embryonic 13.5 fetuses.
Retroviral packaging
The pMX-based retroviral vectors Oct3/4 (Addgene plasmid, 13366), Klf4 (Addgene plasmid, 13370), Sox2 (Addgene plasmid, 13367), and c-Myc (Addgene plasmid, 13375) were obtained from Addgene. For transient transfection, Plat-E cells were transfected with the indicated plasmids using Lipofectamine LTX transfection reagent (Invitrogen). Twenty-four and forty-eight hours after transfection, retroviral supernatant was harvested and filtered into the virus concentration column through a 0.45 μm cellulose acetate filter (Millipore), and they were centrifuged at 4°C, then the virus supernatant was aliquoted and cryopreserved.
Somatic cell reprogramming
To generate the iPSCs, MEFs derived from Daxxfl/fl:Oct4-GFP transgenic and wild-type mice were plated at a density of 50,000 cells per well in 12-well plates and infected overnight with combinations of the pMXs-based retroviral vectors OSKM (Oct4, Sox2, Klf4, and c-Myc) viral supernatant in the presence of polybrene during the first 2 days. Forty-eight hours after transduction, the virus-containing medium was changed. Subsequently, the infected cells were seeded in six-well plates at a density of 20,000 cells per well on the feeder layers and then switched to an iPSC culture medium (knockout Dulbecco's modified Eagle's medium [KO-DMEM], 15% knockout serum replacement [KSR], 1% nonessential amino acids, 1 mM sodium pyruvate, 2 mM
Seven days after transduction, the iPS colonies had smooth edges and defined boundaries. Thirteen days after transduction, colonies were mechanically isolated and further cultured for identification. Then, the iPSCs were examined by both alkaline phosphatase (AP) staining and GFP fluorescence. All animal experiments were performed under the Guidelines for Animal Experiments of the Harbin Medical University (HMUIRB20190011). The methods were approved by the Harbin Medical University Ethics Committee.
Daxx knockout
Daxxfl/fl:Oct4-GFP mouse-derived iPSCs and wild-type iPSCs were infected with Cre lentivirus or empty lentivirus, which both carry the EGFP reporter. The virus was removed, and fresh medium was added 24 hours after infection. Forty-eight hours later, cells were selected using puromycin (2 μg/mL, Solarbio). The medium was changed every other day for 8 days, until a single clone could be recognized under the microscope. The green fluorescent indicator could be observed at 48 hours after selection. At last, the KO efficiency of Daxx was investigated at the RNA and protein levels.
Cell culture
Daxx KO iPSCs and wild-type iPSCs were cultivated on MEF feeder layers in KO-DMEM supplemented with 15% KSR, glutamine, nonessential amino acids, 100 μM 2-ME, and recombinant LIF. The culture dishes were previously coated with 0.1% gelatin. The iPSC colonies were passaged if it becomes bigger, and the growth medium was changed daily. All MEFs and Plat-E (retroviral packaging cell line, maintained culture in our laboratory) were maintained in DMEM containing 10% fetal bovine serum (FBS).
Embryoid body formation
For embryoid body (EB) differentiation, Daxx KO iPSCs and wild-type iPSCs were maintained in embryonic stem cell (ESC) culture medium without feeder cells, induced by withdrawal of LIF, and then cultured on ultralow attachment cell culture dishes (Corning). Pluripotency genes were analyzed by quantitative real-time polymerase chain reaction (qRT-PCR) on different days.
RNA isolation and real-time reverse-transcriptase PCR analysis
The cells were lysed with TRIzol reagent (Invitrogen), and total RNA was purified according to the manufacturer's instructions. One microgram of DNaseI-treated RNA was taken for cDNA synthesis using a PrimeScript RT Reagent Kit (TaKaRa). The qPCR reaction was performed in a Bio-Rad Fast Real-Time PCR System according to the manufacturer's instructions. Primer sequences are listed in Supplementary Table S1.
Alkaline phosphatase staining
Cell colonies were fixed in 4% paraformaldehyde for 2 minutes at room temperature, washed in phosphate-buffered saline (PBS), and incubated in a BCIP/NBT Alkaline Phosphatase Color Development Kit (Beyotime) for 3 hours at room temperature in the dark. After two washes with DPBS, the cell colonies were visualized under a Leica DMI8 inverted microscope.
Immunofluorescence staining
Cells were fixed in 4% paraformaldehyde (Sigma-Aldrich) in PBS at 4°C for 30 minutes. After washing with ice-cold PBS, the cells were blocked in 5% bovine serum albumin (Sigma-Aldrich) with 0.1% Triton at room temperature for 30 minutes. Primary antibodies included Oct4 (1:200, 2840s, Cell Signaling Technology), Sox2 (1:200, 11064-1-AP, Proteintech), and Nanog (1:200, 8822s, Cell Signaling Technology). The secondary antibodies used were CF488 goat anti-mouse IgG (1:500, 20010, Biotium) and CF568 donkey anti-rabbit IgG (1:500, 20098, Biotium). Nuclei were stained with DAPI (Thermo Fisher Scientific). For confocal analysis, the samples were examined with a Nikon laser scanning confocal microscope, and images were acquired using NIS elements analysis software (Nikon).
TUNEL assay
To analyze apoptosis, the TUNEL assay was performed with the one-step TUNEL Kit (C1089, Beyotime) to label the apoptotic stem cells according to the manufacturer's instructions. Briefly, iPSCs were fixed in 4% paraformaldehyde for 30 minutes, then rinsed with PBS and permeabilized in 0.25% Triton X-100 for 5 minutes on ice, followed by the TUNEL assay for 1 hour at 37°C. Cells with red fluorescence were defined as apoptotic cells. Nuclei were stained with Hoechst 33258.
Western blot
Western blot analysis was performed using whole cell extracts prepared by disrupting the cells in SD001 lysis buffer. The protein bands were visualized using an enhanced chemiluminescence reagent after hybridization with a horseradish peroxidase (HRP)-conjugated secondary antibody. Signals were detected with a SuperSignal™ West Pico PLUS Chemiluminescent Substrate for HRP (Thermo Fisher Scientific). All the Western blot results were quantified by ImageJ software.
The antibodies used in this study included anti-Sox2 (1:500, 11064-1-AP, Proteintech), anti-Oct4 (1:500, 2840s, Cell Signaling Technology), anti-Nanog (1:1000, 8822s, Cell Signaling Technology), anti-caspase 3 (1:1000, 19677-1-AP, Proteintech), anti-cleaved caspase 3 (1:500, 9664s, Cell Signaling Technology), anti-Gapdh (1:10,000, KC-5G4, KangChen Bio-tech), β-actin (1:3000, 60008-1-Ig, Proteintech), anti-mouse IgG-HRP (1:3000, ab6728, Abcam), and anti-rabbit IgG-HRP (1:3000, ab6721, Abcam).
Teratoma formation assay
Wild-type and Daxx KO iPSCs were cultured at undifferentiating conditions as described above, harvested by trypsinization, resuspended in DPBS, and injected subcutaneously into both sides of the dorsolateral of immunodeficient mice. Cells (2 × 107) at 200 μL per injection site were used. Teratomas were harvested, fixed, and stained in Hematoxylin and Eosin 6 weeks postinjection.
Neurosphere formation
For iPSC differentiation, Daxx KO iPSCs and wild-type iPSCs were plated onto six-well plates in basic MEM containing 20% FBS (Biological Industries) and 2 mM
RNA sequencing
Total RNA samples were sequenced by BGI genomics technology using BGISEQ, with 20 M reads per sample on an average length of 50 bp. Statistical analysis of the RNA-seq data was used to identify those genes significantly up- or downregulated by treatments, with a false discovery rate <0.05. Fold change was computed with average transcription levels compared with control values, which were in turn log2-transformed and computed for Spearman correlation coefficients between samples.
Statistical analysis
The data were expressed as the mean ± SD of at least three independent samples. Statistical comparisons between groups were performed with a two-tailed Student's t-test; *, p ≤ 0.05; **, p ≤ 0.01; and ***, p ≤ 0.001 were considered significant.
Results
Establishment and characterization of Daxx KO iPSCs
Takahashi and Yamanaka (2006) found that mature somatic cells can be reprogrammed into iPSCs by four pluripotent transcription factors in 2006. In recent years, iPSC technology has exhibited numerous applications; however, the mechanisms of reprogramming are still unclear. To explore the effect of Daxx on self-renewal and differentiation in iPSCs, we selected E13.5 MEFs that were derived from the B6D2F1 mouse strain and the Daxxflox/flox:Oct4-GFP mouse strain (gifted by Prof. Fan Hengyu from Zhejiang University) as donor somatic cells, and then reprogrammed these cells using a retroviral packaging virus of the classical pluripotency transcription factors. Daxxflox/flox:Oct4-GFP fibroblasts exhibited GFP expression when pluripotency genes were switched on in vitro.
Approximately 7 days later, the appearance of the colonies was observed. Then, at day 12, we picked up several Oct4-GFP-positive colonies with typical ESC morphology from both wild-type and Daxx flox iPSCs. These colonies could sustain >10 passages with a highly GFP-positive signal, which illustrates normal Oct4 activity (Fig. 1A).

Establishment and characterization of Daxx KO iPSCs.
To observe the role of Daxx in the iPSCs, we performed KO experiments using lentivirus Cre to infect the Daxx flox iPSCs, and at the same time, we infected the wild-type iPSCs with an empty vector lentivirus carrying the GFP indicator. The expression of green fluorescence was observed at 48 hours after infection, which indicated successful transfection of Cre. We then obtained Daxx KO iPSCs and wild-type iPSCs. To determine the efficiency of KO, we checked Daxx expression by qPCR, which displayed a 1000-fold decrease in Daxx KO iPSCs compared with the wild-type iPSCs at the RNA level (Fig. 1B).
Subsequently, we investigated the expression of Daxx at the protein level, with a western blot, also exhibiting results consistent with RNA expression level (Fig. 1C). An alkaline phosphatase assay demonstrated that the pluripotency activity was maintained in Daxx KO iPSCs (Fig. 1D). Moreover, immunofluorescence staining illustrated a normal expression of the pluripotency genes Oct4 and Sox2 (Fig. 1E). To further identify the differentiation ability of Daxx KO iPSCs, we performed the teratoma formation experiment in vivo. We found that Daxx KO iPSCs were able to form larger teratoma with typical three germ layers (Fig. 1F). This also indicated that iPSCs maintained a normal differentiation ability.
Effect of Daxx deletion on pluripotency and self-renewal
To understand the role of Daxx in miPSCs, we first detected the expression of pluripotency genes, and real-time PCR results demonstrated a higher expression of the pluripotency genes Nanog and Oct4 (Fig. 2A) in Daxx KO iPSCs. Meanwhile, a western blot assay also showed that the expression of Oct4 was slightly elevated (Fig. 2B, C). To further access whether Daxx regulates self-renewal in iPSCs, we performed secondary colony formation assays by replating 1000 wild-type or Daxx KO iPSCs on gelatin for 7 days (Fig. 2D). Soon afterward, as indicated by AP staining, this showed that deletion of Daxx did not impair the colony formation ability of iPSCs (Fig. 2D, E). We hypothesized that this might be relevant to the increased expression of Oct4, which was triggered by Daxx deletion.
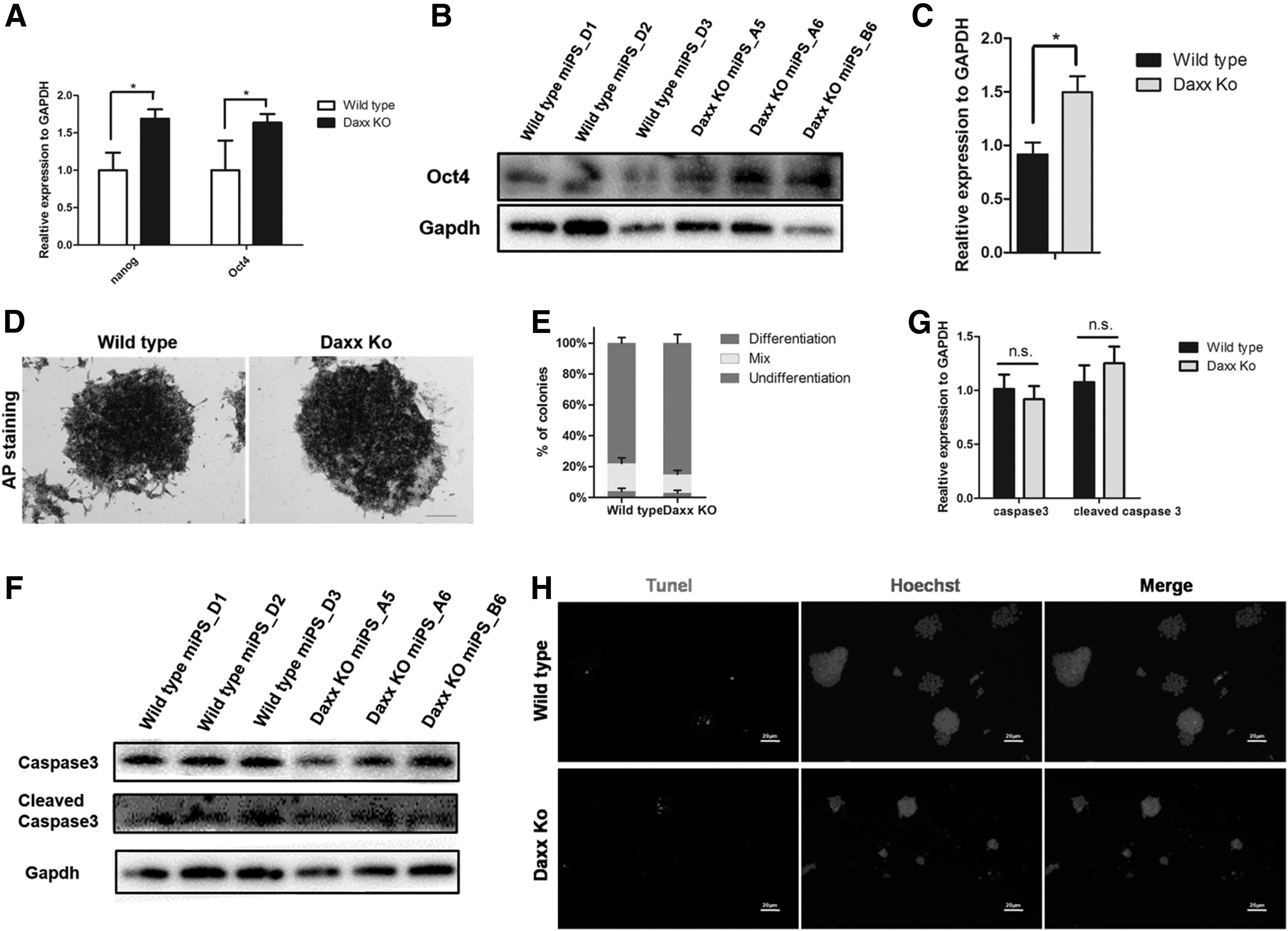
Effect of Daxx deletion on pluripotency and self-renewal
In addition, we found that the expression of endoderm and mesoderm markers had no significant changes. Next, we examined whether Daxx deletion affected cell apoptosis, the expressions of caspase 3 and Cleaved caspase 3 were detected by western blot, and the results illustrated that Daxx deletion would not disturb the expression of apoptotic proteins caspase 3 and cleaved caspase 3 compared with that of wild-type iPSCs (Fig. 2F, G). Meanwhile, we measured cell apoptosis by the TUNEL assay, and found that the number of TUNEL-positive cells in Daxx KO iPSCs was similar to that of the wild-type iPSCs (Fig. 2H). Therefore, we conclude that Daxx deletion does not affect cell apoptosis.
Daxx KO iPSCs displayed differentiation defects
To further investigate the differentiation ability of Daxx KO iPSCs, we performed an EB assay by suspension cultivation without LIF. Twenty-four hours later, EB morphology was observed with smooth borders. As the EBs grew over time, we found that the EBs derived from the Daxx KO group were generally smaller and had a rougher edge than that of the wild-type group (Fig. 3A). Thereafter, we extracted RNA from the EBs from different days and determined the expression of the pluripotency genes Oct4 and Nanog at the RNA level, and it was discovered that Daxx KO EB had a distinctly higher pluripotency gene expression on differentiation days 0, 3, and 4 (Fig. 3B).

Daxx KO iPSCs displayed differentiation defects.
In addition, we performed a 12-day differentiation of EBs to investigate the marker genes of three germ layers by RT-PCR. It was found that the expression level of the ectoderm marker nestin decreased significantly after Daxx KO (Fig. 3C), indicating that the ectodermal differentiation might have been altered. However, the marker genes expression of endoderm and mesoderm did not differ significantly. In brief, it was speculated that Daxx deletion might affect the ectodermal differentiation capacity of iPSCs.
Transcriptome analysis identified differentially expressed genes in iPSCs
To further understand the effects of Daxx deletion on iPSCs, we compared the global transcription profiles in Daxx KO iPSCs relative to their corresponding wild type by RNA transcriptome analysis. Differentially expressed genes were identified by a fold change (FC) >2, and those that were significant in a t-test (p < 0.05). We detected a total of 1921 DEGs that were up- or downregulated in Daxx KO iPSCs relative to the wild-type iPSCs and found that 299 genes were upregulated while 479 genes were downregulated by two-fold in the Daxx KO iPSCs (Fig. 4A). By pathway enrichment analysis, 238 DEGs were shown to be linked to the ribosome biogenesis and rRNA process. Hence, we carried out Gene Ontology (GO) annotation analysis of those altered DEGs in iPSCs using DAVID tools.
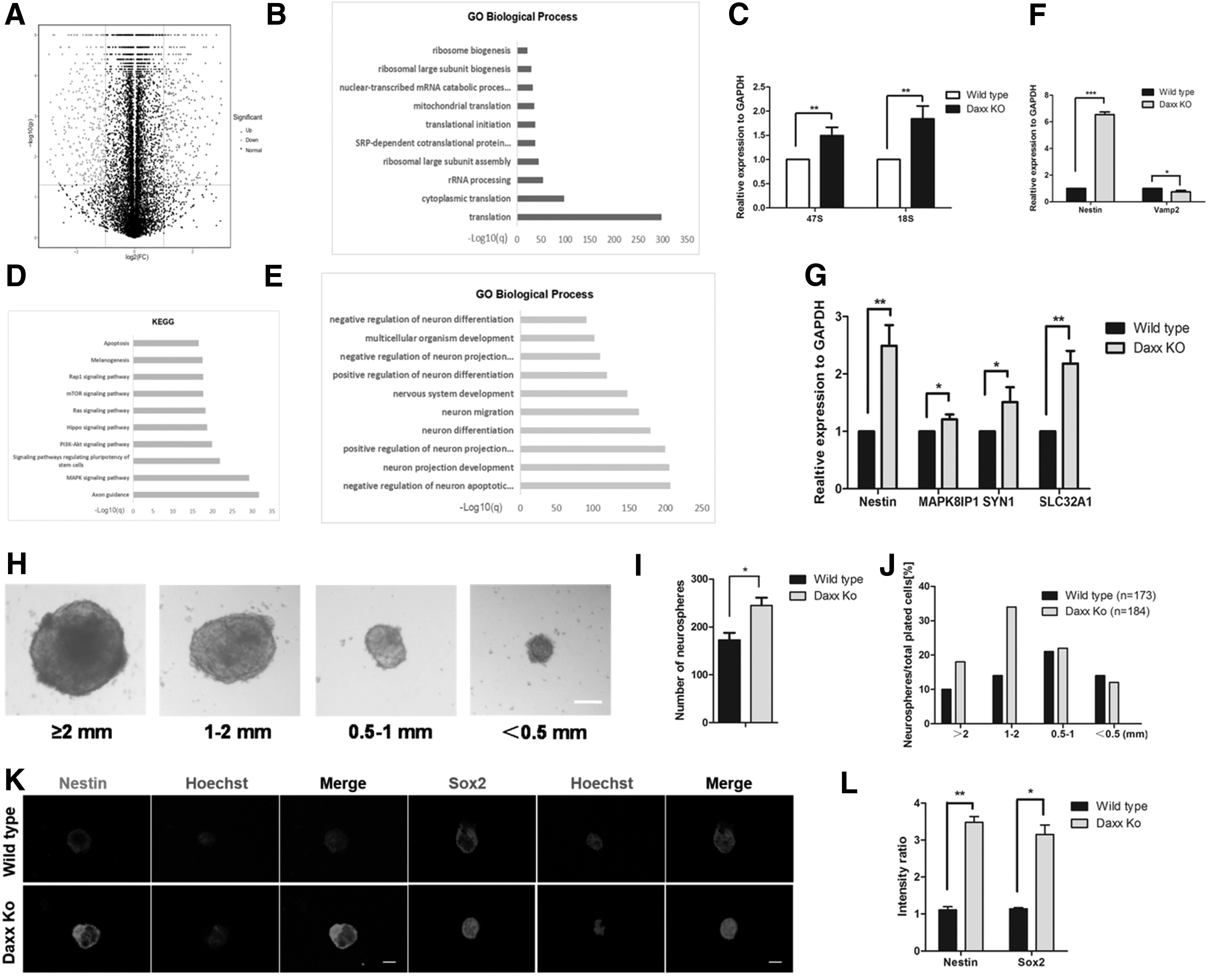
Transcriptome analysis identified differentially expressed genes in wild-type and Daxx KO iPSCs.
The gene ontology terms were significantly enriched in the upregulated genes and were specific to translation, rRNA processing, ribosome large subunit assembly, and biogenesis (Fig. 4B). Simultaneously, qRT-PCR revealed that the expression of rDNA-related genes 47S and 18S was significantly increased (Fig. 4C). We hypothesized that DAXX, as an H3.3 chaperone, contributed less deposition to heterochromatin and caused increased transcriptional activity due to the deletion of Daxx and thus increased the expression of rDNA. Meanwhile, 1538 DEGs were shown to be associated with neural development, and the gene ontology terms significantly enriched in the upregulated gene were specific to neuron projection development and regulation, neuron differentiation, and neuron migration (Fig. 4E).
Following, we examined the expression levels of the neural progenitor gene in iPSCs and found that the expression of nestin had significantly increased (Fig. 4F), indicating that iPSCs were more prone to neural development and differentiation after Daxx deletion. Subsequently, we induced and established the neurospheres from iPSCs. From the data of neurosphere formation, we concluded that the total number of neurospheres was increased in Daxx KO than wild type and the diameter of each neurosphere was measured. A greater number of neurospheres had diameters between 1–2 mm and above 2 mm in Daxx KO. Meanwhile, the smaller size between 0.5–1 mm and below 0.5 mm were similar in Daxx KO and wild type (Fig. 4H–J).
For further evidences, we used the immunofluorescence assay to measure the expression of nestin and Sox2 that were used as markers of neural precursor cells. The results demonstrated expressions of nestin and Sox2 were increased in Daxx KO neuropsheres (Fig. 4K, L). The qPCR results displayed that the expression of nestin was elevated in neurospheres that originated from Daxx KO iPSCs (Fig. 4G). Simultaneously, we performed the KEGG pathway enrichment analysis and found the top pathway terms were enriched in the axon guidance, Mapk signaling pathway, and PI3K-Akt signaling pathway (Fig. 4D).
As shown in Figure 4G, Mapk8lp1, syn1, and SLC32A were overexpressed in the Daxx KO group compared with that of wild type at the RNA level. From the findings above, we speculate that Daxx KO may improve neural differentiation of iPSCs through increasing the expression of related genes.
Discussion
Somatic cells were reprogrammed into pluripotent stem cells by inducing the expression of the transcription factors Oct4/Sox2/c-Myc/Klf4 in 2006 (Takahashi and Yamanaka, 2006). These transcription factors activate gene expression of stem cell-specific genes, regulate their expression, and inhibit genes involved in differentiation. Niwa et al. (2001) found that Oct3/4 is critical for maintaining normal self-renewal of ESCs. In this study, we reprogrammed the MEFs by four pluripotent transcription factors, Oct4/Sox2/c-Myc/Klf4, then we picked up the colonies that established the iPS cell lines. We demonstrated that pluripotency of iPSCs was improved, and expression of the core transcription factors Oct4 and Nanog was increased when knocking out Daxx.
At the same time, we found that the expression of the transcriptional genes Id2, Zfp42, and Tcf15, which are associated with stem cell pluripotency was also increased (Supplementary Fig. S1). Besides, it has been reported that Nanog maintains self-renewal of embryonic stem cells by regulating the expression of downstream target genes gata6 and gata4 (Constantinescu, 2003; Fujikura et al., 2002; Mitsui et al., 2003). In addition, studies have shown that activation of the Wnt signaling pathway could promote the expression of Nanog (Sato et al., 2004). The Wnt signaling pathway may be involved in maintaining ESC pluripotency and self-renewal status (Brandenberger et al., 2004). In this study, we also observed that Wnt signaling pathway members, such as Wnt 3a, Wnt5a, Wnt7b, Wnt9a, and Wnt10b, exhibited elevated levels (Supplementary Fig. S1A).
Meanwhile, we analyzed the expression of the β-catenin and the phosphorylation status of β-catenin by western blot, and it showed downregulation of β-catenin in Daxx KO iPSCs compared with wild-type iPSCs, however, no significant differences in the expression of the phosphorylation status of β-catenin between the two groups (Supplementary Fig. S1B–E). Therefore, we speculate that despite increased expression of wnt, its downstream phosphorylated β-catenin was not activated, suggesting that other pathways may play a role.
Histone components and their covalent modifications determine the state of nucleosomes, which are involved in the epigenetic regulation of chromatin (Buschbeck and Hake, 2017). Histone chaperones prevent promiscuous histone interactions before chromatin assembly, and they ensure that the canonical and functional histone variants are faithfully deposited into chromatin in a spatially and time-limited manner (Zink et al., 2017). ATRX is a chromatin remodeling protein that interacts with its partner DAXX in a replication-independent manner to deposit the histone variant H3.3 in heterochromatin, which plays an important role in gene transcription, epigenetic memory, and reprogramming in the nucleus (Wang et al., 2017; Zink et al., 2017).
In addition, studies have shown that deletion of ATRX in mouse ESCs results in selective loss of the ribosomal RNA gene (rDNA) copy number (Udugama et al., 2018). Then, rDNA copy loss and repeat instability are caused by the destruction of H3.3 deposition, so heterochromatin formation fails in rDNA repeats in the absence of ATRX (Udugama et al., 2018).
At the same time, studies proved that Hira-deficient ESC has the ability to self-renew, but differentiation becomes accelerated after removal of LIF, indicating that H3.3 incorporation is necessary to maintain ESC pluripotency (Meshorer et al., 2006). Considering our experiments, the expression of rDNA-related genes 47S and 18S was improved after Daxx deletion; we hypothesized that the ability of H3.3 deposition in the heterochromatin region of rDNA was reduced after Daxx KO, which acts as a molecular chaperone, and therefore, chromatin accessibility and transcriptional activity were promoted.
Our RNAseq data showed that 1538 differential genes are related to neural function, and Go annotations were mainly enriched in neuron projection, neuron differentiation, and neuron migration. Besides, the KEGG pathway displayed that enrichment was mainly in axon guidance, Mapk signaling, and PI3K-Akt signaling. qPCR also manifested an increased expression of the genes, which were involved in neurodevelopment and neural differentiation. Simultaneously, little has been reported with regard to the connection of Daxx and neural development and differentiation previously. To conclude, our results demonstrated the importance of Daxx for neurodevelopment and neural differentiation; however, further validation is still needed.
Daxx is a multifunctional protein that plays a role in both apoptosis and antiapoptosis (Tang et al., 2004). Previous studies have shown that Axin, P53, and Daxx play a direct role in promoting apoptosis (Li et al., 2007; Wasylishen et al., 2018). In addition, an increase in apoptosis was observed in embryonic stem cells that were knocked out of Daxx, indicating its ability to resist apoptosis (Elsasser et al., 2015; Michaelson et al., 1999). In our experiments, the expression of caspase 3, cleaved caspase 3, and TUNEL assay showed no significant difference between wild-type iPSCs and Daxx KO iPSCs. The apoptotic effect of Daxx on iPSCs needs to be studied further.
Conclusions
In this study, we demonstrated the function of Daxx in iPSCs' pluripotency maintenance and differentiation. Daxx may interact with core pluripotent transcription factors to play a key role in iPSCs. In addition, we indicated the increased expression in rDNA transcription levels and neural development due to the deletion of Daxx. These findings may help us to understand the underlying molecular mechanism of Daxx in reprogramming and pluripotency.
Footnotes
References
Supplementary Material
Please find the following supplemental material available below.
For Open Access articles published under a Creative Commons License, all supplemental material carries the same license as the article it is associated with.
For non-Open Access articles published, all supplemental material carries a non-exclusive license, and permission requests for re-use of supplemental material or any part of supplemental material shall be sent directly to the copyright owner as specified in the copyright notice associated with the article.