
Abstract
Abstract
We previously reported a feeder-free culture method for pure production of subculturable vascular endothelial cells (VECs) from cynomolgus monkey embryonic stem cells (cmESCs) without as using cell-sorting technique. By this method, canonical vascular endothelial (VE)-cadherin/platelet–endothelial cell adhesion molecule 1 (PECAM1)-positive VECs (c-VECs) and atypical VE-cadherin/PECAM1-negative VECs (a-VECs) were generated without a contamination by pericytes, lymphatic endothelial cells, or immature ES cells. More recently, we established a unique culture technique to maintain human ESCs (hESCs) under a feeder-free and recombinant cytokine-free condition. Combining these two systems, we have successfully generated pure VECs from two lines of hESCs, khES-1 and khES-3, under a completely feeder-free condition. Our method is very simple: spheres generated from hESCs by floating culture using differentiation media supplemented with vascular endothelial growth factor, bone morphogenetic protein 4, stem cell factor, FMS-related tyrosine kinase-3 ligand, and interleukin 3 (IL3) and IL6 were cultured on gelatin-coated plates. Cell passage was performed by an ordinary enzymatic treatment. The hESC-derived differentiated cells demonstrated cord-forming activities and acetylated low-density lipoprotein-uptaking capacities. Moreover, they exclusively expressed von Willebrand factor and endothelial nitric oxide synthase. Flow cytometric analyses indicate that khES-3 generated both c-VECs and a-VECs as in the case of cmESCs. By contrast, khES-1 produced only a-VECs, which nonetheless demonstrated effective recruitment into neovascularity in vivo. Interestingly, a-VECs turned to express PECAM1 after transplantation into immunodeficient mice. The hESC-derived VECs were subculturable at least up to 10 passages without functional depression. Our method does not require a presorting processes to enrich progenitor fractions such as CD34-positive or kinase insert domain receptor (KDR)-positive cells, providing the most efficient and easiest technique for VEC production from hESCs.
Introduction
Currently, the methods for vascular endothelial cell (VEC) differentiation of primate ESCs suffer from low differentiation efficiencies (<10%) (Levenberg et al., 2002; Sone et al., 2003, 2007; Wang et al., 2007) and/or difficulties in in vitro expansion (Sone et al., 2007; Sone et al., 2003; et al., 2007; Wang et al., 2007). Moreover, they exclusively require cell-sorting processes to enrich the progenitor populations such as platelet-endothelial cell adhesion molecule 1 (PECAM1) (Levenberg et al., 2002), kinase insert domain receptor (KDR)-positive (Sone et al., 2003, 2007), or CD34-positive (Wang et al., 2007) cells. However, the requirement of cell-sorting processes is disadvantageous in view of technical feasibility. There was one study reporting pure production of subculturable VECs from rhesus monkey ESCs without using a cell-sorting technique (Kaufman et al., 2004). However, these VECs completely lacked the expressions of vascular endothelial-cadherin (VE-cadherin) and PECAM1, the major markers of VECs, although they demonstrated the mature functions in vitro and in vivo. Whether their results can be reproduced in the differentiation of hESCs and whether this uncommon type of VECs, which lacked the message expressions of VE-cadherin and PECAM1 (Kaufman et al., 2004), can justly be recognized as VECs remains undetermined. In addition, all the protocols utilize murine feeder cells for the maintenance culture of hESCs (Levenberg et al., 2002; Sone et al., 2003, 2007; Wang et al., 2007), which is quite unfavorable in view of quality control aiming for clinical application.
Recently, we established a feeder-free culture method for pure production of VECs from cynomolgus monkey ESCs (cmESCs) without progenitor-enriching processes (Saeki et al., 2008). By this method, we could generate VE-cadherin/PECAM1-positive VECs at the efficiency of around 30%. Interestingly, the residual VE-cadherin/PECAM1-negative populations clearly demonstrated the mature functions of VECs: they showed cord-forming activities and acetylated low-density lipoprotein (Ac-LDL)-uptaking capacities in addition to von Willebrand factor (vWF) and endothelial nitric oxide synthase (eNOS) expressions. Moreover, they were negative for monocytic markers including CD14, CD68, and CD45, indicating that the VE-cadherin/PECAM1-negative cells are distinct populations from monocytic VEC progenitors. Although they did not express CD34 or KDR/vascular endothelial growth factor receptor 2 (VEGF-R2), the expression levels of Tie-2 and VEGF-R1 were similar to those of VE-cadherin/PECAM1-positive VECs. Finally, no contamination by pericytes or immature hESCs was observed (Saeki et al., 2008). Based on those findings, we named the VE-cadherin/PECAM1-negative populations generated from cmESCs by our method as “atypical VECs,” which are VE-cadherin−/PECAM1−/CD14−/Tie-2+/vWF+/eNOS+/cord-formation+/Ac-LDL-uptaking+. As we already showed in our report, the key of our success lies in the usage of multiple hematopoietic cytokines in addition to the VEC growth factors (Saeki et al., 2008). Interestingly, no other protocols ever showed the benefit of the usage of hematopoietic cytokines, although hematopoiesis and angiogenesis are intimately associated.
Because our method is superior to other methods in a comprehensive view of efficiency, quality, and feasibility, we planned to examine its applicability to hESCs. In the case of cmESCs, however, the maintenance culture was performed using mouse embryonic fibroblasts (MEFs) (Saeki et al., 2008), which is unfavorable in view of clinical application. Recently, we established a feeder-free and recombinant cytokine-free culture method for the maintenance of hESCs (Nakahara et al., 2009). By combining these two methods, we have succeeded in generating pure VECs, which consists of canonical VECs and atypical VECs, from hESCs under a completely xenogenic cell-free condition without using cell-sorting techniques. Our system provides the nearer way to the clinical application of hES-based or human iPS-based regenerative medicine for vascular disorders than any other methods. In addition, it offers an excellent tool for basic researches on human vascular endothelial biology via an elimination of murine cells throughout the culture process.
Materials and Methods
Cells and reagents
The two lines of hESC (Suemori et al., 2006), khES-1 and khES-3, were cultured by our novel feeder-free and recombinant cytokine-free method (Nakahara et al., 2009). Briefly, cells were subcultured Matrigel™ Basement Membrane Matrix, phenol-Red free (Cat 356237, BD Biosciences, San Jose, CA)-coated culture dishes using DMEM/F12 medium (Invitrogen Corp., Carslbad, CA) supplemented with 20% Knockout Serum Replacement (KSR™) (Invitrogen Corp.), 1% nonessential amino acids solution (Invitrogen Corp.), 1 mM Sodium Pyruvate Solution (Invitrogen Corp.), 2 mM L-glutamine (Invitrogen Corp.), 0.1 mM β-mercaptoethanol (Sigma Chemical Co., St. Louis, MO) 10 U/mL of penicillin (Invitrogen Corp.), and 10 mg/mL of streptomycin (Invitrogen Corp.) without using recombinant cytokines. hESCs were maintained as a crop of colonies of similar sizes with diameters between 200 and 1,000 μm at the densities less than eight colonies per 1 cm2. The ES cells were passed every 4 days. For detachment from the culture dish, cells were treated with a minimum volume of dissociation liquid including 0.25% trypsin (Invitrogen Corp.), 1 mg/mL collagenase IV (WAKO Pure Chemical Industries, Osaka, Japan), 20% KSR™, 1 mM CaCl2 at 37°C for 5 to 15 min. Cells were gently collected without performing pipetting to avoid cell damage. They were seeded at split ratios of 1:2∼1:4 on newly Matrigel™ Matrix-coated dishes, avoiding fusion between colonies. Culture medium was changed every day. Pooled normal human umbilical vein endothelial cells (HUVEC), normal human aortic endothelial cells (HAEC), normal human mesenchymal stem cells (hMSC), normal human aortic smooth muscle cells (AOSMC), and normal human dermal lymphatic microvascular endothelial cells (HMVEC-dLy) were purchased from Lonza Group Ltd., (Basel, Switzerland). The pooled normal human dermal microvascular endothelial cells (HMVEC) and normal human glomerular vascular endothelial cells (HGVEC) were purchased from Dainippon Sumitomo Pharma Co., Ltd. (Osaka, Japan). All the cells were maintained using media recommended by the manufacturers. Murine stromal OP9 cells were maintained with α-MEM medium (Invitorgen Corp.) supplemented with 20% heat-inactivated FBS (PAA Laboratories GmbH), 0.1 mM β-mercaptoethanol (Sigma Chemical Co.), 1 mM L-glutamine (Invitrogen Corp.), 10 U/mL of penicillin (Invitorgen Corp.), and 10 μg/mL of streptomycin (Invitrogen Corp.).
Differentiation procedures
hESCs were detached from culture plates by using the dissociation liquid for 15 min at 37°C. The mildly dissociated ESC clumps were subjected to floating cultures in Hydro cell® (CellSeed Inc., Tokyo, Japan) to form spheres using the differentiation medium consisting Iscove's modified Dulbecco's medium (IMDM) (Sigma Chemical Co.) supplemented with 15% heat-inactivated fetal bovine serum (FBS) (PAA Laboratories GmbH, Linz, Austria), 0.1 mM β-mercaptoethanol, 3 mM L-glutamine, 10 U/mL penicillin, 20 ng/mL vascular endothelial growth factor (VEGFA), 20 ng/mL bone morphogenetic protein 4 (BMP4), 20 ng/mL stem cell factor (SCF), 10 ng/mL FMS-related tyrosine kinase-3 ligand (Flt3-L), 20 ng/mL Interleukin 3 (IL3), and 10 ng/mL IL6. After incubation for 3 days at 37°C under a 100% humidified condition in a 5% CO2 gas incubator, spheres were subjected to adherent culture using 100 × 20 mm 0.1% porcine type A gelatin (Sigma Chemical Co.)-coated dishes in the differentiation medium described above. Media were changed twice a week. For passage, cells were harvested by treatment with 0.25% trypsin and 1 mM EDTA and replated at split ratios of 1:2 on new gelatin-coated dishes. In some experiments, spheres were cultured on 40 Gy-irradiated OP9 feeder layers for control.
Morphological examinations
Viable cells were directly observed under an inverted phase contrast light microscope (Olympus Optical Co. Ltd., Tokyo, Japan).
Cord formation assays
Matrigel™ Basement Membrane Matrix, phenol-Red free (Cat 356237, BD Biosciences) was loaded into the 24 multiwell dishes (95 μL/well). After the dishes were incubated for 30 min at 37°C, 1 × 104 cells per well were seeded in differentiation medium described above. Cell morphologies were observed after overnight culture under an inverted light microscope (Olympus Optical Co. Ltd).
Uptake of acetylated low-density lipoprotein (Ac-LDL)
Cells were transferred in four-well chamber slide system (Nalge Nunc International Corp., Naperville, IL). After overnight culture, cells were washed by Hank's balanced salt solution (HBSS) twice and incubated in serum-free medium containing 10 μg/mL of low-density lipoprotein from human plasma, acetylated, DiI complex (DiI Ac-LDL) (Invitrogen Corp.) for 4 h. After washing the cells by HBSS for three times, nuclei were counterstained using 10 nM of Hoechst 33342 (Sigma Chemical Co.). After washing the cells, samples were observed under the fluorescence microscope (Olympus Optical Co. Ltd).
Flow cytometry
Cells were collected by 0.2% EDTA treatment and, after a wash in phosphate-buffered saline (PBS), 1 × 106 cells were reacted with first antibodies on ice for 30 min. The expression level of each protein was analyzed using a FACSCalibur™ (BD Biosciences). The antibodies used were a mouse monoclonal antihuman VE-cadherin (clone TEA1/31)-phycoerythrin (PE) antibody (Beckman Coulter Inc., Fullerton, CA), a mouse monoclonal antihuman Platelet-Endothelial Cell Adhesion Molecule 1 (PECAM1)-FITC antibody (clone WM59) (BD Biosciences), antihuman Tie-2- allophycocyanin (APC) antibody (R&D Systems Inc., Minneapolis, MN), antihuman VEGF receptor 1 (VEGF R2)-PE antibody (R&D Systems Inc.), antihuman VEGF receptor 2 (VEGF R2)-PE antibody (R&D Systems Inc.), VEGF receptor 3 (VEGF R3)-PE antibody (R&D Systems Inc.), antihuman CD34-PE (clone 356) antibody (BD Biosciences), antihuman CD14-PE (clone M5E2) antibody (BD Biosciences), antihuman CD45-PE antibody (clone TÜ116) (BD Biosciences), or an antihuman SSEA-4-phycoerythrin monoclonal antibody (clone MC813-70, R&D Systems Inc.). After antibody-staining procedures, cells were stained with TO-PRO3 fluorescent dye (Invitrogen Corp.) for 10 minutes. During analysis, dead cells were gated out as the FL4-higher fraction. For analysis on Oct-4 expression, cells were fixed and permeabilized by FIX & PERM® reagents (Invitrogen Corp.) according manufacturer's guidance and stained by an antihuman Oct-3/4-phycoerythrin monoclonal antibody (clone #240408, R&D Systems Inc.).
Immunostaining
The cells were fixed on slide glasses by using a cytospin apparatus (Cytospin 2) along with further fixation with acetone/methanol solution (1:3). The immunostaining procedure was performed as described elsewhere (Saeki et al., 2009) with first antibody reactions using a mouse monoclonal antihuman Oct-3/4 antibody (C-10, Santa Cruz Biotechnology Inc., Santa Cruz, CA), a rabbit polyclonal antihuman Nanog antibody (ReproCELL Inc., Tokyo, Japan), a rabbit antihuman N-cadherin antibody (H-63) (Santa Cruz Biotechnology Inc.), a mouse monoclonal antihuman vascular smooth muscle actin (SMA) (clone 1A4) (Sigma Chemical Co.), a mouse monoclonal antihuman platelet-derived growth factor (PDGF) Receptor β (clone 28) (BD Biosciences), a rabbit polyclonal antihuman eNOS antibody (H-159) (Santa Cruz Biotechnology Inc.), or a goat polyclonal antihuman vWF antibody (C-20) (Santa Cruz Biotechnology Inc.), followed by second antibody reactions using Alexa Fluor® 488 chicken antimouse IgG (H+L), Alexa Fluor® 568 goat antirabbit IgG (H+L) or Alexa Fluor® 594 chicken antigoat IgG (H+L) (Invitrogen Corp.). Nuclear counterstaining was performed using 300 nM of 4′,6-diamino-2-phenylindole (DAPI).
Western blotting
Western blotting was performed as previously described (Saeki et al., 2009) using a mouse monoclonal antihuman calponin 1 antibody (Santa Cruz Biotechnology Inc.), a mouse monoclonal antihuman Oct-4 (C-10) antibody, a rabbit polyclonal antihuman β-tubulin (H-235) antibody (Santa Cruz Biotechnology Inc.), a rabbit antihuman lymphatic vessel endothelial hyaluronan receptor 1 (LYVE-1) antibody (AngioBio Co., Del Mar, CA), a rabbit antihuman prospero-related homeobox 1 (Prox-1) antibody (AngioBio Co.), or a rabbit antihuman Nanog antibody (ReproCELL Inc.). The second antibody reaction was performed using a horseradish peroxidase-conjugated antirabbit or antimouse IgG (Cell Signaling Technology, Inc., Beverly, MA). The final detection procedure was performed using ECL Western blotting detection reagents (GE Healthcare UK Ltd., Buckinghamshire, England).
Matrigel plug assays
The assays were performed as reported by others (Kaufman et al., 2004). Briefly, 3 × 106 of hES-derived differentiated cells were suspended in 10 μL of differentiation medium supplemented with 150 ng/mL of fibroblast growth factor (FGF)-2, mixed with 500 μL of Matrigel™ Basement Membrane Matrix, phenol-Red free (Cat 356237, BD Biosciences) and transplanted subcutaneously into severe combined immunodeficiency (SCID) mice. For control, 10 μL of differentiation medium supplemented with 150 ng/mL of fibroblast growth factor (FGF)-2 was mixed with 500 μL of Matrigel™ Matrix (BD Biosciences) and transplanted in the same way. After 3 weeks, 0.2 mL of fluorescein isothiocyanate (FITC)-dextran (500,000 average molecular size; Sigma Chemical Co.) solution (100 mg was suspended in 5 mL of PBS) was injected to mice from tail veins. After a few minutes, mice were sacrificed and the blocks were fixed by 10% formaldehyde and paraffin embedded. The 4-μm sliced specimen were directly observed under fluorescent microscopy to determine the presence of FITC–dextran-packed lumens in the pugs. They were then subjected to immunostaining studies using a rabbit polyclonal antihuman PECAM1 antibody (H-300) (Santa Cruz Biotechnology Inc.), which does not recognize murine vascular endothelial cells as we showed in Figure 4C, or mouse monoclonal anti-HLA-A, B, C antibody (BD Biosciences).
Adipocyte differentiation
Confluent cultures of hMSCs and hES-derived cells in six-well plates were subjected to adipocyte differentiation as follows. First, cells were cultured with hMSC Differentiation BulletKit-Adipogenic (Lonza Walkersville, Inc., Walkersville, MD) for 3 days. Then, medium was changed to MSCGM BulletKit (Lonza Walkersville, Inc.). After overnight culture, medium was replaced by hMSC Differentiation BulletKit-Adipogenic. Then, the cells were cultured for 3 days, when medium was changed to MSCGM BulletKit. After another overnight culture, cells were fixed and subjected to oil red O staining at day 8 of the differentiation culture.
Oil red O staining
Cells were washed with PBS twice and fixed by 10% formalin solution for 10 min at room temperature. After washing with PBS twice, cells were incubated in 60% isopropanol solution for 1 min. Then, cells were stained by Oil Red O Stain Original Solution (Muto Pure Chemicals Co. Ltd, Tokyo, Japan) for 20 min at room temperature. After rinsing with 60% isopropanol solution and with PBS, cells were directly observed under phase contrast microscopy.
Karyotype analyses
The metaphase hESCs were collected after 16-h incubation of hESCs with 0.06 μg/mL of colcemid. ES cells were treated by hypotonic buffer and fixed by methanol/acetic acid (3:1). Chromosomal analyses with Giemsa band (G-band) staining were performed by SRL, Inc. (Tokyo Japan) by a standard method.
Results
Pure production of canonical VECs and atypical VECs from hESCs
We recently established a feeder-free culture method for pure production of subculturable VECs from cynomolgus monkey embryonic stem cells (cmESCs) that were maintained on murine feeder layers. To further advance this technique toward clinical application, we tried VEC differentiation of hESCs that were maintained by our novel feeder-free, recombinant cytokine-free culture method (Nakahara et al., 2009).
By adding a minor modification to the original protocol, we successfully induced VEC differentiation of two lines of hESCs, khES-1 and khES-3. First, we generated spheres from hESCs by a simple floating culture using the differentiation medium supplemented with hematopoietic cytokines (Fig. 1A). Then, hESC-derived spheres were cultured on gelatin-coated plates. As in the case of cmESCs (Saeki et al., 2008), cobblestone-shaped cells spread from the detached spheres (data not shown) and we transferred them massively onto new gelatin-coated plates after enzymatic treatments. Cells were actively proliferated and reached confluence (Fig. 1B). Compared to the case of cmESC (Saeki et al., 2008), hESC-derived cells were rather morphologically heterogeneous. However, this is also the case with the primary culture of human vascular endothelial cells: some areas of HUVEC showed compact cobblestone appearance consisting of smaller cells, whereas other areas showed rather loose images consisting of larger cells (Fig. 1B, right). Although there is a quantitative difference: khES-1-derived cells mainly consisted of larger loose-contacted cells; khES-3-derived cells consisted of smaller compact cells and larger loose-contacted cells; HUVEC consisted of mainly smaller compact cells, it can be said that there was no morphologically crucial differences among the three types of cells.
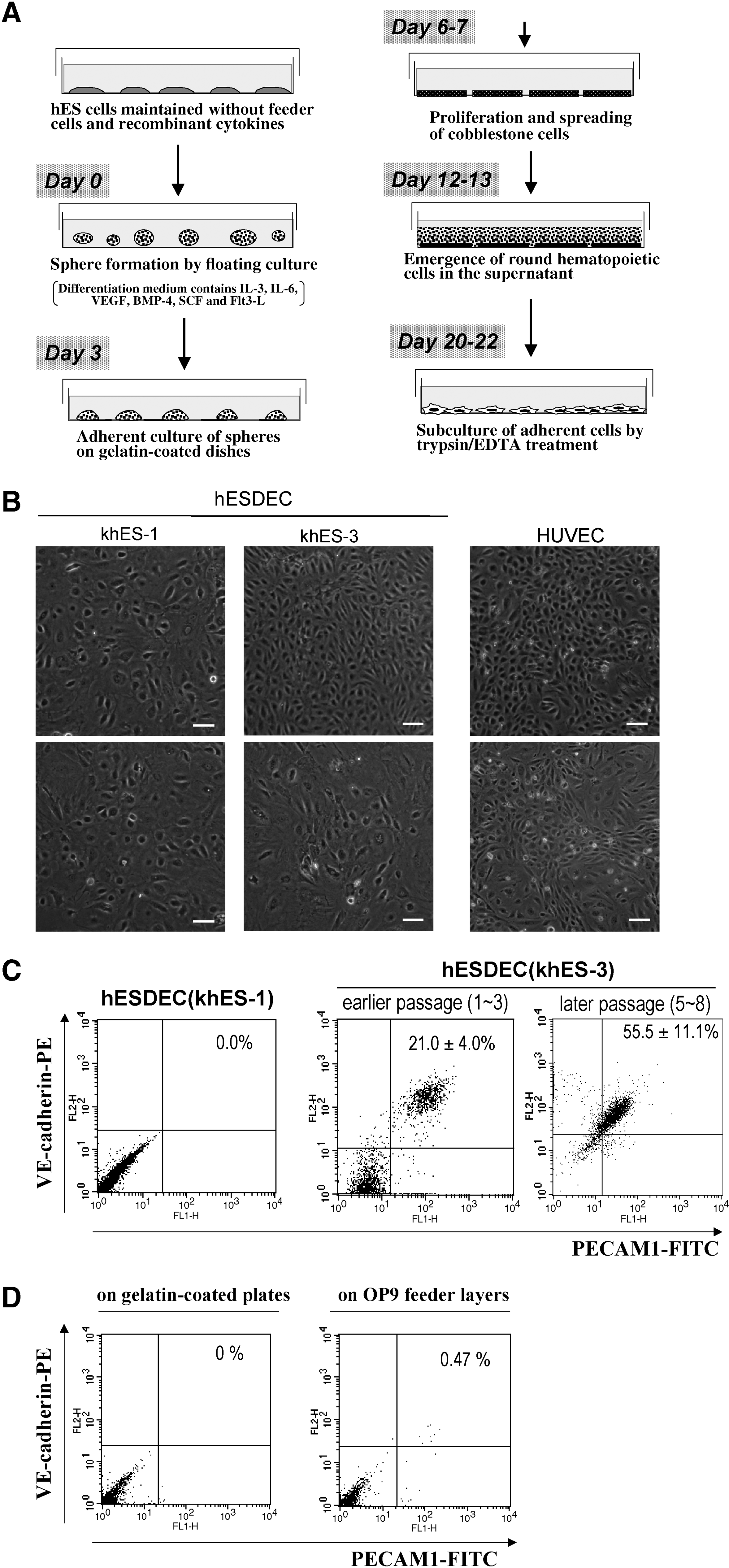
The VEC differentiation of khES-1 and khES-3. (
To evaluate the performance of VEC differentiation, we examined the cell surface expressions of VE-cadherin and PECAM1 by flow cytometry. As in the case of cmESCs, about 20% of the khES-3-derived cells was VE-cadherin/PECAM1-positive, whereas all the khES-1-derived cells were VE-cadherin/PECAM1-negative (Fig. 1C). Concerning khES-3 line, percentages of VE-cadherin/PECAM1-positive populations did not decrease but rather increased during the course of subculture by unknown reasons (Fig. 1C, right). On the other hand, khES-1 cells scarcely produce VE-cadherin/PECAM1-positive populations even in the presence of OP9 feeder layers (Fig. 1D).
We next examined the possibility that the khES-1-deirved cells as well as the khES-3-derived VE-cadherin/PECAM1-negative-populations were a-VECs. As shown in Figure 2A and B, both of the khES-1-derived cells and khES-3-derived cells showed cord-forming activities and uniform Ac-LDL-uptaking capacities. They exclusively expressed vWF and eNOS (Fig. 2B). We found no significant differences in N-cadherin protein expressions between the khES-1-derived and khES-3-derived cells. Despite the absence of VE-cadherin protein expression in khES-1-derived cells as demonstrated by flow cytometry (Fig. 1C), VE-cadherin message expression was detected by RT-PCR although its expression levels were considerably declined with an increment in passage number (Fig. 2D).

Characterization of hESC-derived VECs. (
We further examined the expressions of other VEC markers including VEGF receptors, Tie-2, and CD34 in comparison with primary cultures of various human VECs including human glomerular endothelial cells (HGVEC), human dermal microvascular endothelial cells (HMVEC), human aortic endothelial cells (HAEC), and human umbilical vein endothelial cells (HUVEC). Tie-2 expression levels in khES-1-derived and khES-3-derived cells were higher than those of HMVEC and HGVEC. On the other hand, VEGF R2 expression was detectable only in minor populations of hESC-derived cells as shown by histogram analyses (Fig. 2E). Nevertheless, we could detect low-level expressions of VEGF R2 in majority of khES-1-derived and khES-3-derived cells by dot plot analyses (Fig. 2F). CD34 was absent in entire populations of khES-1-derived cells and in major parts of HGVEC, HMVEC, and HAEC, whereas it was expressed in about half populations of HUVEC and khES-3-derived cells. The expression levels of VEGF R1 were similar among all kinds of VECs (10–30%), while substantial variations were found in VEGF R3 expressions levels (10–70%). In general, hES-derived cells express various VEC markers, and variations in the expression levels of those markers between khES-1-derived and khES-3-derived cells were not significantly larger than those among human VECs derived from different tissues.
We next checked the contamination of hES-derived VECs by other lineage cells. Cogeneration of pericytes was ruled out by the absence of a mature smooth muscle cell marker of calponin-1 (Fig. 3A), a smooth muscle cell/macrophage maker of actin α-2 (Fig. 3B) and pericyte/mesenchymal stem cell marker of PDGF-Rβ (Fig. 3C). Coexistence of lymphatic endothelial cells was excluded by the absence of LYVE-1 and Prox-1 (Fig. 3D). Possibility that VE-cadherin/PECAM1-negative populations would be monocytic VEC progenitors was also excluded by the absence of a monocyte marker of CD14 (Fig. 3E) and a pan-leukocyte marker of CD45 (Fig. 3F). Contamination by immature hESCs was ruled out by the absence of immature hESC markers of Nanog (Fig. 3G), and Oct3/4 (Fig. 3G) as shown by Western blotting. This finding was further supported by flow cytometric analyses on SSEA-4 expressions: there was no significant differences between the isotype IgG3 control-staining pattern and the anti-SSEA-4 antibody-staining pattern in both khES-1-derived and khES-3-derived cells as demonstrated by histogram (Fig. 3H) and dot plots (Fig. 3I). Finally, the presence of mesenchymal stem cell-like populations was ruled out by the lack of adipocyte differentiation potentials of hES-derived cells (Fig. 3J). All these findings together indicate that the khES-1-derived cells and khES-3-derived VE-cadherin/PECAM1-negative cells are a-VECs.

Exclusion of contamination by other lineage cells. (
We also checked the chromosomal stability of the hESC-derived cells. As shown in Fig. 3K, no chromosomal abnormalities were detected in khES-1-derived cell or khES-3-derived cells, at least by karyotyping studies with G-banding.
Phenotypes of hESC-derived cells were stable during the subculture process: cord-forming activities and Ac-LDL-uptaking capacities at passage 10 (Fig. 3L) were comparable to those at passage 4 (Fig. 2A). Moreover, no significant morphological changes were observed until the cells entered senescence, when they became enlarged and flattened (data not shown). hESC-derived VECs were easily expanded by regular passages twice a week by 1:2 dilution. Accordingly, we could obtain 2 × 109 VECs from 6 × 106 hESCs after 10 passages.
From all these results, we concluded that our feeder-free differentiation method enabled pure production of VECs, which include c-VECs and a-VECs, from hESCs that were maintained under a feeder-free condition without inducing chromosomal abnormalities.
Evaluation of in vivo functions of a-VECs
Although the in vitro studies have confirmed the functional maturation of a-VECs, which bear cord-forming activities and Ac-LDL-uptaking capacities (Fig. 2A) and express vWF (Fig. 2B), eNOS (Fig. 2B), N-cadherin (Fig. 2C), and Tie-2 (Fig. 2E), it is a crucial point whether they indeed demonstrate neovascular formation activities in vivo if we consider hESC-derived VECs as a tool of regenerative medicine.
To address this issue, we performed Matrigel plug assay using khES-1-derived a-VECs. As shown in Figure 4A, transplantation of khES-1-derived cells resulted in an effective formation of neovascularity that were linked with the host circulation system (Fig. 4A, lower). By contrast, transplantation of sole Matrigel did not induce a neovascular formation (Fig. 4A, upper). To confirm the recruitment of khES-1-derived cells into neovascularity, we determined the presence of human cells by immunostaining studies using an antihuman HLA-A, -B, -C antibody. We found that the majority of the cells lining at the neovascularity expressed human antigens as shown in a tangentially sectioned sample (Fig, 4B). The absence of the human HLA-A, -B, -C-staining in some areas (arrows in Fig. 4B, left middle) might be due to the recruitment of host murine VECs, which inevitably occurs during the connection between the khESC-1-derived neovascularity and the host circulation system. Unexpectedly, majority of the neovescularity-constituting cells were stained by human PECAM1 antibody (Fig. 4C, top panels). Because this antibody specifically recognizes human VECs and does not react with murine VECs (Fig. 4C, forth panelS from the top), it was suggested that a-VECs turned to express PECAM1 after transplantation. On the other hand, neovascular tissues were scarcely stained by anti-SMA antibody (Fig. 4C, third panels from the top), indicating that murine pericytes were not efficiently recruited into the neovascularity. Although this finding possibly raises an issue of impaired maturation of neovascularity, it at least guarantees that a-VECs would not transformed into pericytes even after transplantation. By contrast, VE-cadherin expression was not detected even after transplantation (data not shown).
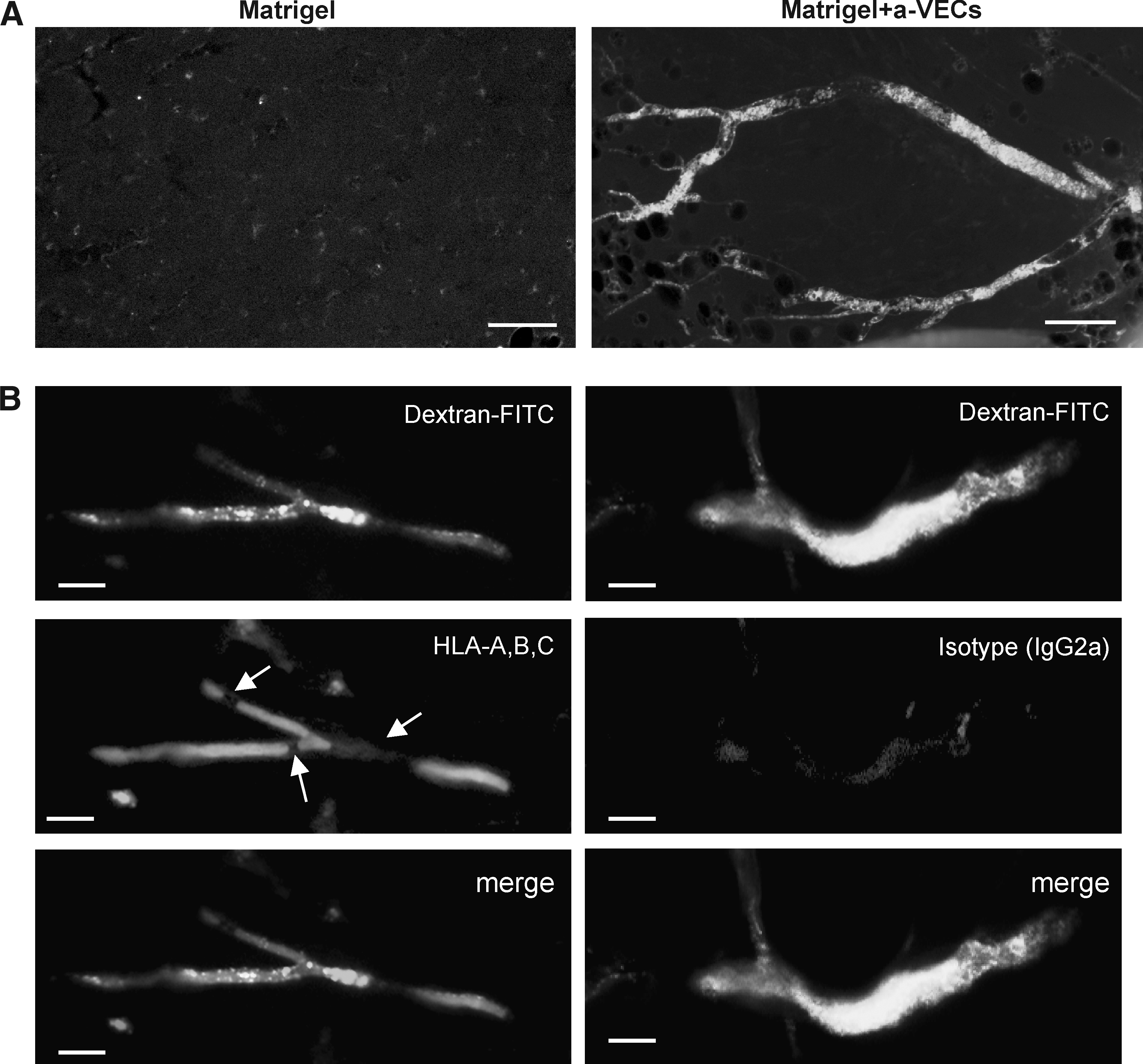
Evaluation of neovascularity-forming activities of a-VECs in vivo. Matrigel plug assays. Matrigel plug assays were performed using khES-1-derived cells at passage 11 to passage 14 as described in Materials and Methods. (
Thus, hESC-derived a-VECs were functional in vivo, demonstrating effective recruitment into neovascularity in SCID mice.
Discussion
We reported our success in producing pure VECs from hESCs. Our system has three advantages. First, the process is completely free from xenogenic cells including an immature hESC-maintaining step. Second, it does not require cell-sorting techniques. Finally, the product does not contain immature hESCs or other lineage cells including pericytes and lymphatic endothelial cells. In our knowledge, there is no other method that satisfies all those conditions.
Our method had originally been established using cmESC. To apply this method to hESCs, we added a slight but technically critical modification to the original protocol. For sphere formation, cmESCs were incubated by a hanging drop culture. Although hanging drop culture has an advantage in generating homogenous sized spheres, it induced substantial damages to hESCs. So, we tried simple floating culture using low attachment plates. Despite a considerable heterogeneity in sphere sizes, we successfully accomplished VEC differentiation of khES-3 with a comparable quality to cmESCs. Due to its higher feasibility, floating culture method has enabled us to handle larger amounts of hESCs at a time. The additional improvement is that we used the hESCs maintained under feeder-free conditions as starting materials, which is an advantage in view of clinical application.
By our system, both cell surface VE-cadherin/PECAM1-positive c-VECs and VE-cadherin/PECAM1-negative a-VECs were generated from primate ESCs. There is a large variation in the ratio of c-VECs to a-VECs among ESC lines: both cmESCs and khES-3 produces 20∼30% of c-VECs, whereas khES-1 produced only a-VECs. Eventually, khES-1 scarcely produced c-VECs even on OP9 feeder layers (Fig. 1D). As for now, we do not know the reason for this variation. Recently, it was reported that there were marked differences in differentiation propensity among hESC lines (Osafune et al., 2008). Thus, an intrinsic character of each ESC line may affect the quality of VEC differentiation. As we reported previously, both cmESCs (Nakahara et al., 2008) and khES-3 (Saeki et al., 2009) effectively produced hematopoietic cells including functional neutrophils while khES-1 cells do not (Saeki et al., 2009). Common molecular signals are involved in the development of hematopoietic and endothelial cells (Lugus et al., 2005). It is widely accepted that adult hematopoietic cells are principally derived from VE-cadhein-positive hemogenic endothelial cells in mice (Lancrin et al., 2009; Taoudi et al., 2008), and the presence of hemogenic endothelial cells was also reported in human (Wu et al., 2007). As we showed in studies on cmESCs, hematopoietic cytokines have beneficial effects for an effective VEC differentiation (Saeki et al., 2008). From all those findings, we speculate that the hESC lines suited for hematogenesis are advantageous in producing c-VECs. Further studies will illuminate the molecular basis for the differentiation propensity.
Whether a-VECs have in vivo counterparts is an interesting and important issue. Our preliminary data indicate that primary cultures of human VECs including HUVEC and aortic endothelial cells (HAEC) contain minor fractions (<5%) of VE-cadherin-negative and/or PECAM1-negative populations, although the percentages of those fractions vary among lots. Eventually, a-VECs turned to express PECAM1 within 3 weeks after transplantation as we showed in Matrigel plug assays (Fig. 4D), suggesting that certain in vivo microenvironments induced the switching towards c-VECs. It might be possible that c-VECs and a-VECs could be interchangeable, depending on their microenvironments. Alternatively, there might be a spectrum between a-VECs and c-VECs. Further investigations are required for the evaluation of in vivo relevance of a-VECs.
Our preliminary data indicate that our differentiation method is applicable to human iPS cells (Gokoh, M., in preparation), providing a way to the realization of order-made regenerative medicine for vascular disorders. Before clinical application, however, the differentiation procedure must be improved to meet with the requirement of Good Manufacturing Practice. At present, we use following xenogenic materials: KSR™ and Matrigel™ Matrix during the maintenance culture of hESCs; FBS and porcine gelatin during the differentiation process. We have found that Matrigel™ Matrix can be substituted by a mixture of type IV collagen and laminin derived from human placenta (Nakahara, M., unpublished observations), and that FBS can be replaced by KSR™ although the expansion rates of VECs are diminished (Yogiashi, Y., and Gokoh, M., unpublished observations). Replacement of KSR™ by human serum and/or synthetic materials should be further tried in aim of clinical application.
Footnotes
Author Disclosure Statement
The authors declare no conflicting financial interests exist.