
Abstract
Background:
This study aimed to determine the efficacy and safety of pioglitazone/glimepiride as a fixed-dose combination (FDC) in Japanese patients with type 2 diabetes.
Subjects and Methods:
In this multicenter, Phase III, open-label evaluation, eligible patients had to have a glycosylated hemoglobin (HbA1c) level of ≥7.4% and <10.4% halfway through a 4-week run-in period while being treated with glimepiride 1 or 3 mg once daily plus diet and exercise. At baseline, patients were assigned to 8 weeks of treatment with pioglitazone/glimepiride (15 mg/1 mg) FDC once daily (Group A; n=31) or pioglitazone/glimepiride (30 mg/3 mg) FDC once daily (Group B; n=31) according to their glimepiride dose during run-in.
Results:
Pioglitazone/glimepiride significantly reduced the mean HbA1c level from baseline (primary end point) by 0.59±0.556% in Group A (P<0.0001) and by 0.55±0.637% in Group B (P<0.0001). Corresponding reductions in the mean fasting blood glucose level were 12.5±21.67 mg/dL (P=0.0032) and 29.1±35.38 mg/dL (P<0.0001). Significant alterations from baseline to week 8 in either one or both treatment groups were also noted for the following parameters: 1,5-anhydroglucitol, glycoalbumin, triglycerides, high-density lipoprotein cholesterol, and free fatty acid levels. Five patients in Group A (16.1%) had five treatment-related adverse events, and 10 patients in Group B (32.3%) had 13 such events; all events were mild.
Conclusions:
Pioglitazone/glimepiride as a FDC (30 mg/3 mg and 15 mg/1 mg once daily) significantly improved glycemic control and lipid profiles and was well tolerated in Japanese patients with type 2 diabetes.
Introduction
A general trend throughout clinical therapeutics is for broader use of single-pill fixed-dose combinations (FDCs). 5,7 In hypertension, for example, a wide range of FDC preparations is now available worldwide, and diabetes has followed suit. That is, various fixed-dose antidiabetes preparations (e.g., glyburide/metformin, pioglitazone/metformin, and, more recently, pioglitazone/glimepiride) are commercially available in the global setting, with the fundamental goals being to improve patient convenience, treatment adherence, and glycemic control and to reduce diabetes management costs. 4,5,7,8 It is important that a recent systematic review of treatment adherence, satisfaction, and costs in individuals with type 2 diabetes treated with a single-pill FDC rather than a “loose pill” combination regimen revealed that adherence was 10–13% greater in patients treated with FDCs, that patients generally preferred FDCs as shown from satisfaction assessments on the Diabetes Treatment Satisfaction Questionnaire, and that patients treated with FDCs had a lower use of healthcare resources and lower direct monthly healthcare costs. 8
Concomitant use of pioglitazone, which improves insulin resistance, and glimepiride, which promotes insulin secretion, 9 is a well-established approach to treating patients with type 2 diabetes. In 2006–2007, FDC tablets comprising pioglitazone/glimepiride at respective dosages of 30 mg/2 mg, 30 mg/4 mg, and 45 mg/4 mg once daily were approved in Western countries. In Japan, combination therapy with these two constituents, administered concomitantly but not in the same tablet, was first approved more than a decade ago, and 15 mg/1 mg and 30 mg/3 mg formulations of a pioglitazone/glimepiride FDC became available in 2011.
The current multicentre, open-label, Phase III trial was conducted to determine the efficacy and safety of two dosages of the pioglitazone/glimepiride FDC (30 mg/3 mg and 15 mg/1 mg once daily) in Japanese patients with type 2 diabetes.
Subjects and Methods
This was a multicenter, Phase III, open-label, 8-week evaluation of a new fixed-dose pioglitazone/glimepiride combination tablet in Japanese patients with type 2 diabetes who were currently being treated with glimepiride plus diet and exercise. The study was performed at five centers in Japan between September 2008 and February 2009 and in accordance with ethical principles set out in the Declaration of Helsinki and International Conference on Harmonisation Tripartite Guidelines for Good Clinical Practice. The study was approved by Institutional Review Boards at each study site, and all subjects provided written informed consent to participate.
The trial involved a 4-week run-in period and an 8-week treatment period. Enrolled patients with type 2 diabetes had to be ≥20 years old, with a glycosylated hemoglobin (HbA1c) level of ≥7.4% and <10.4% at 2 weeks after the start of the run-in period (week −2); this was despite receiving a stable treatment regimen of glimepiride 1 or 3 mg once daily, plus specific dietary and exercise therapy, during the 4 weeks before the start of the run-in period.
The main exclusion criteria were as follows: administration of any antidiabetes drug (including insulin) other than glimepiride within 4 weeks of, or during, the run-in period; previous treatment with a thiazolidinedione agent; patients with hepatic or renal impairment, serious cardiovascular, pancreatic, or hematological disorders, gastrointestinal disorders, such as diarrhea and/or vomiting, or any malignancy; drug (including alcohol) abuse/dependency; hypersensitivity to glimepiride or sulfonamides; pregnant or lactating women; and women of child-bearing age not using adequate contraception.
Each subject maintained his or her glimepiride dosage (1 or 3 mg once daily orally) during the 4-week run-in period at the same level as was administered during the 4 weeks before run-in. During the 8-week treatment period, patients previously receiving glimepiride 1 or 3 mg once daily were switched to pioglitazone/glimepiride 15 mg/1 mg (Group A) or pioglitazone/glimepiride 30 mg/3 mg (Group B) once daily orally, respectively. Patients were instructed to maintain dietary and exercise therapy at the same levels throughout the study.
During the run-in period, the following parameters were assessed and recorded: demographics/patient characteristics, physical examination, vital signs, medical history, body weight, body mass index, 12-lead electrocardiogram (repeated on weeks 0, 4, and 8), clinical laboratory tests, concomitant medications, pregnancy test, HbA1c, fasting blood glucose (FBG), glimepiride dosage, compliance with diet/exercise, and adverse events (AEs).
At baseline (week 0) and after 2, 4, and 8 weeks of treatment, recorded parameters included physical examination, vital signs, clinical laboratory tests, body weight, concomitant medications, compliance with diet/exercise, and AEs. Measures of glycemic control, such as HbA1c, FBG, fasting insulin, 1,5-anhydroglucitol (1,5-AG), and glycoalbumin, as well as fasting serum lipid levels, were recorded at weeks 0, 2, 4, and 8. Plasma samples were taken to determine concentration measured at the end of a dosing interval (C trough) levels of unchanged glimepiride and its metabolites (M1 [hydroxylated glimepiride] and M2 [carboxylated glimepiride]) at weeks 0 and 2. All week 8 assessments were performed at the end-of-treatment visit.
Outcome measures
The primary study end point was the change from baseline in the HbA1c level. The value for HbA1c (%) was estimated as a National Glycohemoglobin Standardization Program (NGSP) equivalent value (%) calculated according to the formula HbA1c (%)=1.02×HbA1c (Japanese Diabetes Society) (%) +0.25%, considering the relational expression of HbA1c (Japanese Diabetes Society) measured by previous Japanese standard substance and measurement methods and HbA1c (National Glycohemoglobin Standardization Program). 10 The main secondary study end point was the change in the FBG level during the course of the study. Other secondary end points included changes in fasting insulin, glycoalbumin, 1,5-AG, and fasting serum lipid levels (total cholesterol [TC], triglycerides [TGs], high-density lipoprotein-cholesterol [HDL-C], low-density lipoprotein-cholesterol [LDL-C], and free fatty acids [FFAs]). All laboratory assessments were performed at an independent, central laboratory.
Safety measures
Safety was assessed by recording AEs, vital signs, 12-lead electrocardiogram, clinical laboratory test results, and body weight. AE reporting included investigator assessments of severity and the relationship of an AE to study drug.
Statistical methods
The planned sample size was 52 patients: 26 in each treatment group. Efficacy assessments were performed for the full analysis set, which included all patients who received at least one dose of study medication during the run-in period and who satisfied the study inclusion criteria. For the primary and secondary end points (HbA1c and FBG, respectively), summary statistics and corresponding two-sided 95% confidence intervals (CIs) were calculated by treatment group at all time points. Mean changes and two-sided 95% CIs from baseline to weeks 2, 4, and 8 and end of treatment were also calculated, and a one-sample t test was performed. For statistical testings, the significance level was at 0.05 (two-sided).
Safety assessments included all patients who received at least one dose of study medication. Safety findings were summarized using descriptive statistics or frequency distributions.
Results
In total, 70 patients were enrolled in the study and assigned to receive either pioglitazone/glimepiride 15 mg/1 mg (Group A; n=36) or pioglitazone/glimepiride 30 mg/3 mg (Group B; n=34) once daily. Of these, five patients in Group A and three patients in Group B did not satisfy the inclusion criteria and were ineligible for study participation beyond the run-in phase; these patients did not receive the FDC tablet. Thus, in total, 62 patients (Group A, n=31; Group B, n=31) completed the 8-week treatment period and comprised the full analysis set (Table 1) for the primary end point (change in HbA1c level from baseline). As one patient in Group B was excluded from the population because of poor treatment adherence (<80%) during the run-in period, the per-protocol population comprised 61 patients (Group A, n=31; Group B, n=30) (Fig. 1).
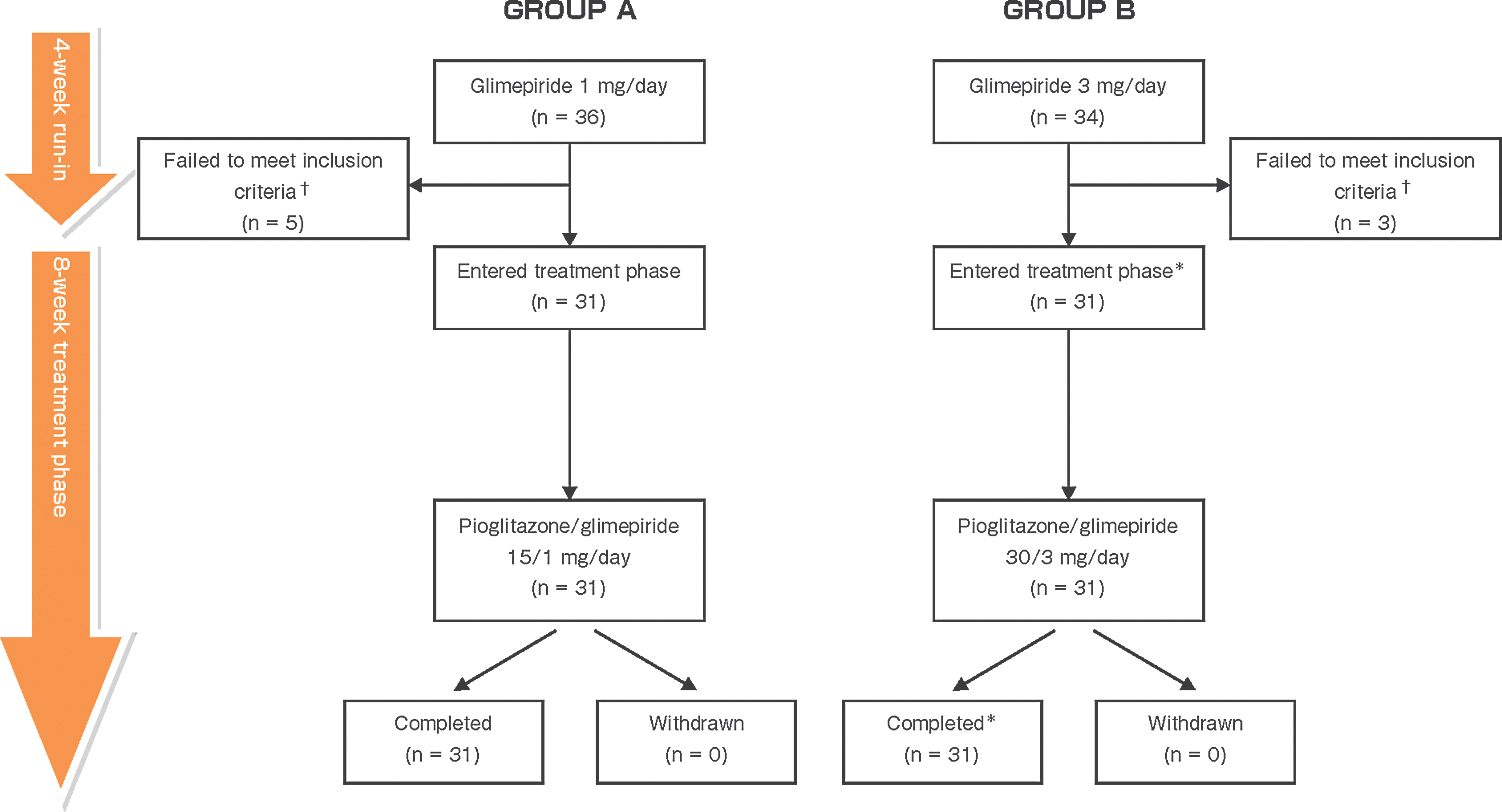
Patient disposition. †Mainly because the glycosylated hemoglobin level was outside the required range of ≥7.4% and <10.4% at 2 weeks after the start of the run-in period. *The full analysis set included 31 patients from Group B, whereas the per-protocol set included only 30 patients (i.e., one patient was excluded because of poor treatment adherence [<80%] during run-in).
Data are expressed as mean±SD values unless otherwise indicated.
1,5-AG, 1,5-anhydroglucitol; FBG, fasting blood glucose; HbA1c, glycosylated hemoglobin; TC, total cholesterol.
No major differences in baseline characteristics were noted between the two treatment groups (Table 1). Overall, approximately two-thirds of patients were male, the mean±SD duration of diabetes was about 7.6±6.24 years, and approximately one-quarter of the total population had diabetes complications, hypertension, and/or hyperlipidemia. Baseline mean values for HbA1c, FBG, and fasting total cholesterol were 8.59±0.749%, 178.4±35.72 mg/dL, and 217.0±33.85 mg/dL, respectively. The mean duration of exposure to pioglitazone/glimepiride during the treatment period was 57.2±2.48 days in Group A and 56.4±1.78 days in Group B. The treatment adherence rate in both groups during the 8-week treatment period was 99.1%.
Primary study end point
Pioglitazone/glimepiride significantly reduced mean HbA1c level from baseline: by 0.59±0.556% in Group A (P<0.0001) and by 0.55±0.637% in Group B (P<0.0001) (Fig. 2). The actual change in the mean HbA1c level in Group A was from 8.36% at baseline to 7.78% at week 8; the corresponding change in Group B was from 8.81% to 8.26%. Reductions were significant at all study time points except at week 2 in the higher-dose pioglitazone/glimepiride group. In the per-protocol analysis, an HbA1c decrease of 0.59±0.556% was recorded in the 15 mg/1 mg group (P<0.0001), and a decrease of 0.54±0.646% was recorded in the 30 mg/3 mg group (P<0.0001).
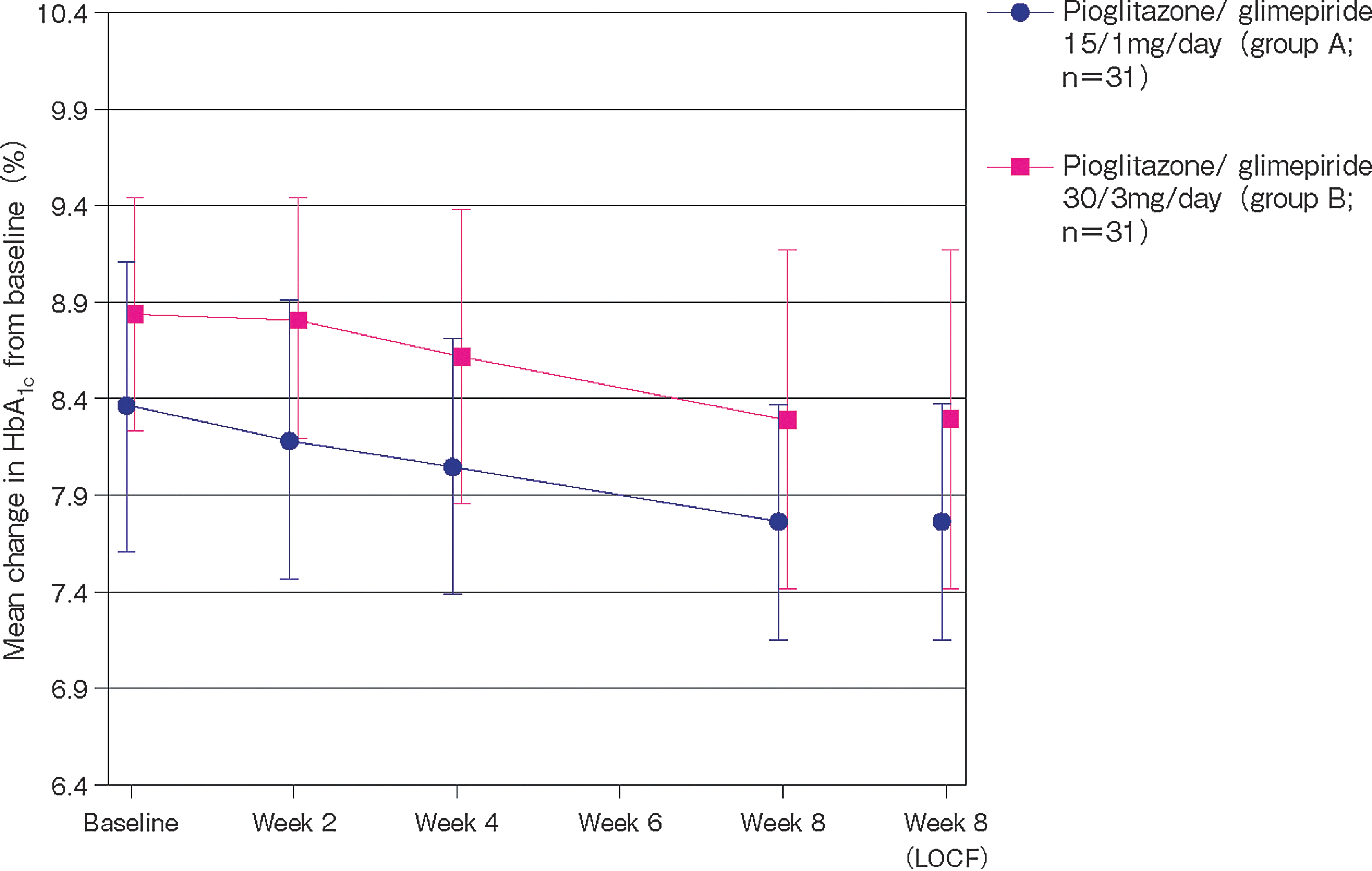
Mean change in glycosylated hemoglobin (HbA1c) levels from baseline to week 8 (primary study end point). LOCF, last observation carried forward. Regarding significance, P<0.0001 for all time points after baseline, except for week 2 in Group B (not significant) and week 4 in Group B (P=0.0061).
Secondary study end points
Pioglitazone/glimepiride significantly reduced mean FBG level from baseline: by 12.5±21.67 mg/dL in Group A (P=0.0032) and by 29.1±35.38 mg/dL in Group B (P<0.0001) (Fig. 3). The actual change in the mean FBG level in Group A was from 166.0 mg/dL at baseline to 153.5 mg/dL at week 8; the corresponding change in Group B was from 190.8 mg/dL to 161.6 mg/dL. Reductions were significant at all time points.
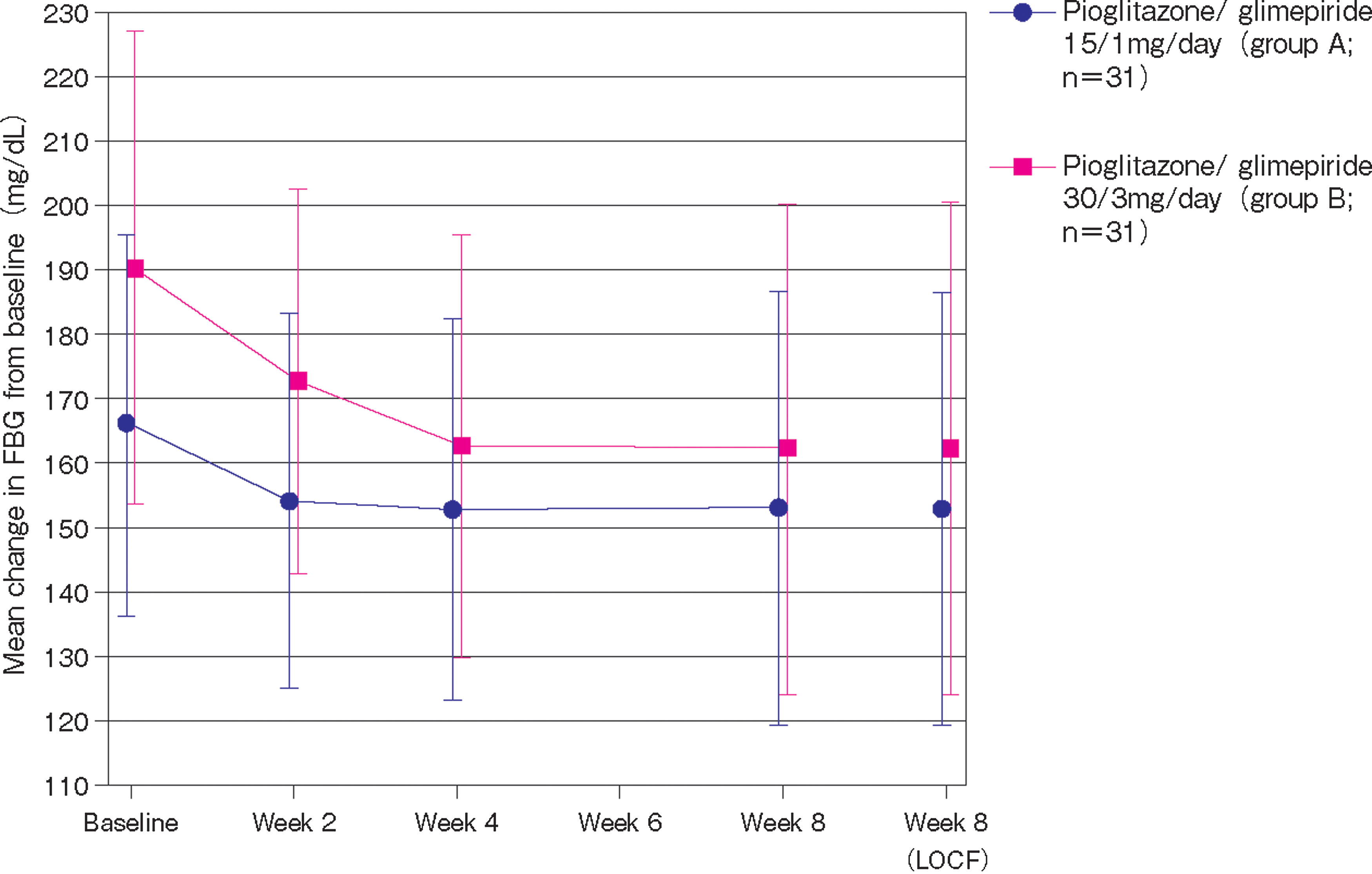
Mean change in fasting blood glucose (FBG) from baseline to week 8. LOCF, last observation carried forward. Regarding significance, values were statistically significant at all time points: all P≤0.0036 in Group A; all P<0.0001 in Group B except at week 2 (P=0.0013).
In Group A, the fasting insulin level was not reduced significantly from baseline at week 8 (−0.70 μU/mL; P=0.444), although reductions at week 2 (−1.52 μU/mL; P=0.0059) and at week 4 (−1.68 μU/mL; P=0.0147) were significant. In Group B, reductions in the fasting insulin level were significant at all time points and ranged between −1.81 to −2.57 μU/mL during the 8-week observation period.
In both the lower-dose (Group A) and higher-dose (Group B) groups, pioglitazone/glimepiride produced significant changes from baseline to week 8 in levels of various other secondary efficacy parameters (Table 2): 1,5-AG, +22% and +54%, respectively; glycoalbumin, −7% and −8%, respectively; fasting TGs, −21% with the higher dose; fasting HDL-C, +6% and +16%, respectively; and FFAs, −15% and −24%, respectively. However, the combination tablet at either dose had no significant effects on levels of TC and LDL-C.
Data are mean±SD values. Group A received pioglitazone/glimepiride 15 mg/1 mg/day (n=31); Group B received pioglitazone/glimepiride 30 mg/3 mg/day (n=31).
1,5-AG, 1,5-anhydroglucitol; FFA, free fatty acid; HDL-C, high-density lipoprotein-cholesterol; LDL-C, low-density lipoprotein-cholesterol; NS, not significant; TC, total cholesterol; TG, triglyceride.
C trough values for glimepiride and its M1 and M2 metabolites at week 2 of the treatment phase were similar to values at week 0 (i.e., after administration of glimepiride 1 or 3 mg once daily for up to 4 weeks), suggesting no effect of pioglitazone on the pharmacokinetic profile of glimepiride.
Safety outcomes
No patient discontinued study treatment because of AEs, and no patient had serious AEs. The incidence of AEs considered possibly, probably, or definitely related to treatment was 16.1% (five of 31) in Group A and 32.3% (10 of 31) in Group B (Table 3). All treatment-related AEs were considered mild in severity. Overall, the most frequent treatment-related AEs were edema (one patient in Group A; four patients in Group B) and increased body weight (n=2 and 3, respectively). Among the five patients with increases in body weight, actual increases ranged from 3.7 to 4.4 kg (from 4.5% to 6.6%) over 8 weeks. In the entire full analysis set, mean body weight increased by 1.3 kg (from 65.1 kg) in Group A and by 2.1 kg (from 69.1 kg) in Group B. Only one patient, who was treated with pioglitazone/glimepiride 30/3 mg daily, experienced hypoglycemia; the event was mild, and the patient recovered.
Data are number (%) values.
AE, adverse event; BNP, brain natriuretic peptide; CPK, creatine phosphokinase.
Discussion
This multicenter, open-label trial demonstrates that, in Japanese patients with type 2 diabetes, 8 weeks of therapy with pioglitazone/glimepiride FDCs (either 15 mg/1 mg or 30 mg/3 mg once daily) significantly improves glycemic control relative to glimepiride monotherapy as indicated by favorable changes in HbA1c, FBG, 1,5-AG, and glycoalbumin and significantly enhances lipid profiles as shown by favorable alterations in TGs, HDL-C, and FFAs. A dose correlation was observed between high-dose and low-dose groups for the change in FBG from baseline to study end. Had the administration period of pioglitazone/glimepiride FDCs been longer than 8 weeks, it is expected that a dose-dependent decrease in HbA1c would also have been evident.
These results corroborate earlier data outlining therapeutic advantages for pioglitazone plus sulfonylurea combination therapy, relative to sulfonylurea treatment alone, in individuals with type 2 diabetes. 11 –13 In the randomized PROspective pioglitAzone Clinical Trial In macroVascular Events (PROactive) study, approximately 1,000 patients received pioglitazone (or placebo) plus a sulfonylurea for a mean follow-up period of almost 3 years. 11 The regimen comprising pioglitazone plus sulfonylurea, as opposed to sulfonylurea alone, was associated with a significantly greater decrease in mean HbA1c (−0.9% vs. −0.4%; P<0.001), a greater proportion of patients attaining HbA1c goals (64.1% vs. 38.9% at HbA1c<7.0%; P<0.001), and considerably fewer patients progressing to permanent insulin use (6.3% vs. 14.8%; P<0.001). 11
Furthermore, a large-scale observational study involving 2,388 individuals with type 2 diabetes showed that 12 months of treatment with pioglitazone plus a sulfonylurea versus metformin plus a sulfonylurea produced significantly greater changes in HbA1c (−1.53% vs. −0.97%; P<0.001), HDL-C (+8.3% vs. +4.3%; P<0.001), LDL-C (−15.2% vs. −11.3%; P<0.001), and TGs (−20.7% vs. −15.2%; P<0.001). 12 Finally, a randomized controlled study of pioglitazone as add-on therapy to a sulfonylurea, which involved 560 patients treated for 16 weeks, reported significant reductions in mean HbA1c (−0.8% to −1.2%) and mean FBG (−33.8 mg/dL to −52.3 mg/dL) levels with the pioglitazone plus sulfonylurea combination compared with the placebo plus sulfonylurea combination. 13
Relative to the individual constituents being administered concurrently but separately, FDC formulations are associated with marked clinical benefits such as reduced pill burden, improved patient convenience and satisfaction, and improved treatment adherence. These benefits, in turn, may provide additional improvements in glycemic control and reduced diabetes management costs. 3 –7 In the current trial, levels of treatment adherence to pioglitazone/glimepiride schedules were high (>99%), and the FDC was well tolerated: all reported treatment-related AEs were mild.
Although the pioglitazone/glimepiride FDCs showed good results in this trial, the findings need to be interpreted with caution as the study has some limitations. Principally, the trial was of an open-label design, included only small numbers of patients, and was not placebo-controlled. Although the objective measures used to evaluate efficacy can overcome these limitations to some extent, additional well-designed, large-scale, randomized, double-blind studies would be appropriate to better define any efficacy, safety, economic, and quality-of-life advantages for pioglitazone/glimepiride FDC over “traditional” dosage schedules. Similarly, well-designed, direct head-to-head comparisons of pioglitazone/glimepiride with other established and emerging antidiabetes FDCs would be useful to delineate more clearly the relative place of specific FDCs in evidence-based guidelines for the treatment of type 2 diabetes.
An increased risk of incident bladder cancer with pioglitazone use has been reported in various studies and meta-analyses, 14 –17 but the association has generally been relatively weak. As the patient population exposed to pioglitazone tends to have more advanced and complicated diabetes, the apparent increased risk may reflect at least to some degree the presence of multiple independent risk factors for bladder cancer. 18 The final report of a 10-year cohort study conducted in the General Practice Research Database in the United Kingdom indicates no significant increased risk of bladder cancer with pioglitazone compared with other antidiabetes drug treatments. 19 This finding is mirrored by the most recent interim (8-year) analysis of the ongoing 10-year Kaiser Permanente Northern California cohort study. 20 Notwithstanding, until such time as sufficient data accumulate to conclude definitively whether or not pioglitazone is a promoter for urinary bladder tumors, prescribing instructions recommend that it not be used in patients with active bladder cancer. In patients with a history of bladder cancer, the benefits of improved glycemic control with pioglitazone need to be weighed against the unknown risks for cancer recurrence. (Note that these are prescribing instructions for pioglitazone and not the views of the authors.)
Pioglitazone/glimepiride FDC is already approved for the treatment of type 2 diabetes in several countries worldwide, and the current study extends the evidence for its superiority over sulfonylurea monotherapy to Japanese patients. In diabetes management, the use of multidrug schedules containing agents with complementary modes of action continues to increase. The use of FDCs such as pioglitazone/glimepiride, which has favorable effects on glycemic and lipid profiles as well as providing convenience of administration, is expected to increase in line with this general trend. 5,7
Conclusions
This multicenter, open-label study showed that pioglitazone/glimepiride FDCs at dosages of 15 mg/1 mg and 30 mg/3 mg once daily significantly improved glycemic control and lipid profiles and were well tolerated in Japanese patients with type 2 diabetes.
Footnotes
Acknowledgments
The authors thank the following investigators for their assistance with this trial: Dr. Koutaro Shimokawa, Yutenji Internal Medicine, Tokyo, Japan; Dr. Hajime Onda, Tokyo Clinical Research Organization for Medicine Clinic, Tokyo; Dr. Yasuyuki Enomoto, Nihonbashi Enomoto Internal Medicine, Tokyo; Dr. Haruna Azuma, Haruna Clinic, Tokyo; Dr. Shigeto Kanada, Heishinkai Medical Group Inc. OCROM Clinic, Osaka, Japan; and Dr. Ryotaro Sasaki, Sasaki Clinic, Amagasaki, Japan. Fixed-dose pioglitazone/glimepiride 15/1 mg and 30/3 mg tablets were provided by the CMC Center, Pharmaceutical Technology R&D Laboratories, Takeda Pharmaceutical Company Ltd., Osaka. This clinical trial was funded by Takeda Pharmaceutical Company Ltd.
Author Disclosure Statement
S. Hiroi, K. Sugiura, K. Matsuno, M. Hirayama, and K. Kuriyama are employees of Takeda Pharmaceutical Company Ltd. K. Kaku has acted as a consultant for Takeda, Astellas, Novo Nordisk, Sanwa, and Novartis and has received research funding from AstraZeneca, Daiichi-Sankyo, Dainippon-Sumitomo, MSD, Novartis, Novo Nordisk, Sanofi-Aventis, Sanwa, and Takeda. K. Kawakami has no competing financial interests. S. Hiroi is responsible for the concept/design and drafting of the article. K. Sugiura is responsible for data analysis and statistics. K. Matsuno is responsible for pharmacokinetics/pharmacodynamics analysis. M. Hirayama is responsible for pharmacokinetics/pharmacodynamics analysis and statistics. K. Kuriyama is responsible for drafting of the protocol. K. Kaku is responsible for clinical approval of the article. K. Kawakami is responsible for data interpretation.