
Abstract
Vesicular stomatitis virus (VSV) replication is highly sensitive to interferon (IFN)-induced antiviral responses. Pretreatment of sensitive cultured cells with IFNβ results in a 104-fold reduction in the release of infectious VSV particles. However, differences exist between the mechanisms of reduced infectious particle titers in cell lines of neuroblastoma and nonneuronal lineage. In L929-fibroblast-derived cells, using immunofluorescence confocal microscopy, infection under control conditions reveals the accumulation of VSV matrix, phosphoprotein (P), and nucleocapsid (N) proteins over time, with induced cellular morphological changes indicative of cytopathic effects (CPEs). Upon observing L929 cells that had been pretreated with IFNβ, neither detectable VSV proteins nor CPEs were seen, consistent with type I IFN antiviral protection. When using the same techniques to observe VSV infections of NB41A3 cells, a neuroblastoma cell line, aside from similar viral progression in the untreated control cells, IFNβ-treated cells illustrated a severely attenuated VSV infection. Attenuated VSV progression was observed through detection of VSV matrix, P, and N proteins in isolated cells during the first 8 h of infection. However, by 18–24 h postinfection all neuroblastomas had succumbed to the viral infection. Finally, upon closer inspection of IFNβ-treated NB41A3 cells, no detectable changes in VSV protein localization were identified compared with untreated, virally infected neuroblastomas. Next, to extend our study to test our hypothesis that virion assembly is compromised within type I IFN-treated neuroblastoma cells, we employed electron microscopy to examine our experimental conditions at the ultrastructural level. Using VSV-specific antibodies in conjunction with immuno-gold reagents, we observed several similarities between the two cell lines, such as identification of viroplasmic regions containing VSV N and P proteins and signs of stress-induced CPEs of VSV-infected cells, which had either been mock-treated or pretreated with interferon-β (IFNβ). One difference we observed between nonneuronal and neuroblastoma cells was more numerous actively budding VSV virions across untreated L929 plasma membranes compared with untreated NB41A3 cells. Additionally, IFNβ-treated, VSV-infected L929 cells exhibited neither cytoplasmic viroplasm nor viral protein expression. In contrast, IFNβ-treated, VSV-infected NB41A3 cells showed evidence of VSV infection at a very low frequency as well as small-scale viroplasmic regions that colocalized with viral N and P proteins. Finally, we observed that VSV viral particles harvested from untreated VSV-infected L929 and NB41A3 cells were statistically similar in size and shape. A portion of VSV virions from IFNβ-treated, virally infected NB41A3 cells were similar in size and shape to virus from both untreated cell types. However, among the sampling of virions, pleomorphic viral particles that were identified from IFNβ-treated, VSV-infected NB41A3 cells were different enough to suggest a misassembly mechanism as part of the IFNβ antiviral state in neuroblastoma cells.
Introduction
IFNs are a family of proinflammatory cytokines classified into three types. Type I IFNs are composed of a broad range of homologs, including IFNα and IFNβ; type II contains only IFNγ; type III is comprised of a family of related IFNλs (Pestka et al., 2004). IFNα/β and IFNγ are the most abundant and immunological-relevant species (Muller et al., 1994; van den Broek et al., 1995). Type I IFNs are directly induced by viral infections
IFNs signal through heterodimeric receptors, leading to activation of receptor-mediated tyrosine kinases that translocate into the nucleus to initiate transcription at either IFN response elements or IFNγ-activated sites (GAS). IFN response elements and GAS-driven expression yield the myriad of IFN-stimulated gene (ISG) products responsible for establishing an antiviral state within which viral infection is retarded and/or blocked completely (Goodbourn et al., 2000; Platanias, 2005).
The activation of several ISGs have been well characterized in response to viral infections of nonneuronal cells, for example, double-stranded RNA-dependent protein kinase R and 2′,5′-oligoadenylate synthetase (Goodbourn et al., 2000). Alternative IFN-induced antiviral pathways also exist that create a proapoptotic environment (e.g., induction of caspases), physically obstruct virion release (e.g., tetherins and Mx GTPase family proteins), modify viral RNA (e.g., induction of double-stranded RNA-dependent adenosine deaminase and APOBEC family proteins), or disrupt assembly and budding (e.g., ISG15-ylation targeted interference of mono-ubiquitination) (Wong et al., 1977; Maheshwari et al., 1980; Goodbourn et al., 2000; Argyris et al., 2007; Neil et al., 2008; Okumura et al., 2008; Sadler and Williams, 2008).
Owing to its exquisite sensitivity to IFN-induced antiviral pathways, vesicular stomatitis virus (VSV) is one of several viruses that have been used to study IFN responses both in vitro and in vivo (Huang et al., 1993; Muller et al., 1994; van den Broek et al., 1995; Durbin et al., 1996; Ireland and Reiss, 2006; Trottier et al., 2007; Detje et al., 2009). VSV is a member of the Rhabdoviridae viral family, and is most notable for its bullet-shaped morphology, insect vector transmission, and its impact on commercial livestock (Letchworth et al., 1999; Rose and Whitt, 2001). VSV possesses a relatively small, negative-sense RNA genome encoding the five viral proteins necessary for replication, assembly, and budding of its enveloped virions. The VSV proteome consists of the following: nucleocapsid (N), phosphoprotein (P), matrix (M), glycoprotein (G), and the large subunit. The P and large subunit proteins assemble to form the viral RNA-dependent RNA polymerase (RDRP) (Perlman and Huang, 1973; Emerson and Yu, 1975; De and Banerjee, 1984, 1985; Hwang et al., 1999). The N protein protects the viral genome and facilitates RDRP function through formation of a ribonucleoprotein (RNP) complex (Masters and Banerjee, 1988; Gupta and Banerjee, 1997), which is either directed or diffuses through the cytosol toward the plasma membrane before assembly. At the plasma membrane, RNP, along with small amounts of RDRP, is encapsidated by M, while nascent virus buds through membrane microdomains containing G (Jayakar et al., 2004).
Studies have shown that type I IFNs are induced in mice infected intravenously, intraperitoneally, or intranasally with VSV, leading to effective clearance of the pathogen from the periphery of immunocompetent hosts (Huang et al., 1993; Muller et al., 1994; van den Broek et al., 1995; Trottier et al., 2007). Disruption of the type I IFN pathway results in severe host compromise and rapid death from VSV infection (Huang et al., 1993; Muller et al., 1994; Durbin et al., 1996; Detje et al., 2009). Intranasal VSV infection of immunocompetent hosts leads to encephalitis without type I IFN production within the CNS, even though it is readily produced in peripheral lymphoid tissues at 24 hours postinfection (hpi) (Ireland and Reiss, 2006; Trottier et al., 2007; Okumura et al., 2008). Type I IFN present in the periphery is unable to cross the blood–brain barrier and inhibit VSV replication in the CNS (Dafny and Yang, 2005). No induction of IFN expression was found in studies of VSV-infected primary neurons ex vivo or neuroblastoma cell lines in vitro (Trottier et al., 2005). However, when these cells are pretreated with IFNβ before VSV infection, a profound attenuation in the release of infectious particles was observed—an abrogation largely independent of any inhibition to viral translation, transcription, and viral genomic replication (Trottier et al., 2005; D'Agostino et al., 2009). Further, VSV infection of neurons in the presence of IFNβ and specific inhibitors of well-characterized IFN-dependent antiviral effector pathways (e.g., protein kinase R) has no effect on the efficacy of IFN treatment, indicating suppression of viral replication by other pathways (Trottier et al., 2005).
Recently, we have shown that VSV infection of L929 and NIH/3T3 cells, widely studied murine tissue culture cell lines, pretreated with IFNβ resulted in a 104-fold reduction in the release of infectious particles with a concomitant abrogation in viral transcript and protein levels. However, in cell lines of neuronal lineage, we found only a 3-fold reduction in viral transcript and proteins levels, despite the same 104-fold reduction in released infectious virions. These data suggested an assembly defect resulting from a type I IFN-induced antiviral state. Further analysis of potential posttranslational modification events revealed IFNβ-related alterations in M and P protein phosphorylation (D'Agostino et al., 2009). Hypophosphorylation of VSV P protein was found to occur in neuroblastoma cell lysates, but not within budded virions from the same IFNβ-treated cells. In contrast, hyperphosphorylated VSV M protein was observed in both cell lysates and viral particles from IFNβ-treated neuroblastoma cells. In addition, hyperphosphorylated VSV M protein was found to inhibit its associations with the N protein and may further suggest a misassembly mechanism in type I IFN treated in neuroblastoma cells (D'Agostino et al., 2009).
Given our previous observations in neuroblastoma cells, as well as published findings in other cell types, we sought to identify phenomena consistent with viral misassembly using confocal immunofluorescence microscopy and electron microscopy. Using both murine NB41A3 neuroblastomas and L929 fibroblast/adipocyte-derived cell lines, VSV infection was monitored over time to examine spatial or temporal changes in viral protein localization patterns. In addition, using electron microscopy in conjunction with VSV-specific antibodies and immuno-gold reagents, we further examined viral protein localization at the ultrastructural level. The identification of aberrant protein localization, in addition to the observed biochemical changes in viral proteins, could provide further evidence for a defect in VSV assembly and point toward a particular mechanism employed by IFNβ-induced neuroblastoma cells.
Materials and Methods
Cells lines, virus, and general reagents
NB41A3 neuroblastoma cells (ATCC CCL-147, Manassas, VA) and L929 (ATCC CCL-1) were cultured as previously described (Trottier et al., 2005). VSV Indiana strain, San Juan serotype, was originally obtained from Alice S. Huang (then at The Children's Hospital, Boston, MA). Murine IFN-β (R&D Systems, Minneapolis, MN), bovine fibronectin (Sigma-Aldrich, St. Louis, MO), Triton X-100 (Sigma-Aldrich), Tween-20 (Sigma-Aldrich), blocking agent (Blotto; BioRad, Hercules, CA), and Aclar film (Allied Signal Specialty Films, Pottsville, PA) were used. Further, ammonium acetate (Sigma-Aldrich), bovine serum albumin (Sigma-Aldrich), ethylenediaminetetraacetic acid (EDTA; Sigma-Aldrich), amyl acetate (Sigma-Aldrich), paraformaldehyde (Electron Microscopy Sciences, Hatfield, PA), glutaraldehyde (Electron Microscopy Sciences), London Resins (LR) White medium set (Electron Microscopy Sciences), uranyl acetate (Electron Microscopy Sciences), sodium cacodylate (Electron Microscopy Sciences), and osmium tetroxide (OsO4; Electron Microscopy Sciences) were also used.
Tissue was cultured using a complete medium of either F12K (Mediatech, Herndon, VA) or Dulbecco's modified Eagle's medium (Mediatech) supplemented with donor horse serum (Mediatech) and/or fetal bovine serum (Atlanta Biologicals, Lawerenceville, GA), in addition to other cell-line-specific supplements cited by ATCC. Lastly, viral plaque assays were performed as previously described (Trottier et al., 2005).
Time-course infections of neuroblastoma and nonneuronal cells for immunofluorescence staining
Aclar film was cut to size to fit inside 30 mm polystyrene tissue culture plates (Becton Dickinson, Franklin Lakes, NJ). After sterilization with two 30 min washes with 70% ethanol, Aclar pieces were washed once with sterile phosphate-buffered saline (PBS) and treated with 50 μg/mL sterile bovine fibronectin for 30 min to 1 h. After aspiration of the fibronectin solution, NB41A3 or L929 cells were immediately plated with 1 × 105 cells per plate in the cell culture medium appropriate for each cell line. The following day, half of the cells were treated with 400 U/mL IFNβ and the other half treated with vehicle alone; cells were subsequently incubated for an additional 16–24 h within tissue culture incubators.
VSV was always added to cells in serum-free media, at a multiplicity of infection equal to 3, and allowed to adsorb for 30 min at 37°C, 5% CO2. Subsequently, unbound virus was removed by washing the monolayers several times with sterile Hank's balanced salt solution (Mediatech) and re-fed with complete media for the duration of the infection. From 3 hpi and continuing hourly until 8 hpi, VSV released into supernatants was harvested for plaque assays. The remaining cells on Aclar membranes were gently washed with excess PBS and fixed for 20 min at room temperature with a 3% paraformaldehyde (Electron Microscopy Sciences) + 0.5% glutaraldehyde (Electron Microscopy Sciences) + PBS solution. After fixation the cells were washed again with excess PBS and any remaining fixative was neutralized by 20 min incubation at room temperature with 1 M glycine + PBS. After one final wash with excess PBS, cells were stored in PBS at 4°C.
Immunofluorescence staining for confocal microscopy
Aclar films with fixed, VSV-infected cells, from both control and IFNβ treatment groups, were cut down to size using surgical scissors and placed into wells of Lab TeK II 8-chamber tissue culture slides (Becton Dickinson). These fixed cell sections were then permeabilized by 3 min incubation at room temperature with 0.2% Triton-X100 + PBS, followed by a wash with excess PBS. Fixed cell sections were then blocked with 3% Blotto + 0.5% Tween-20 for 30 min at 37°C, followed by primary antibody treatment in 3% Blotto + 0.5% Tween-20 with 1:100 rabbit anti-VSV P antiserum (a gift from Gail Wertz) (Green et al., 2000), 1:100 rabbit anti-VSV N antiserum (a gift from Gail Wertz) (Green et al., 2000), or 20 μg/mL mouse anti-VSV M 23H12 monoclonal antibody (a gift from Doug Lyles) (Lefrancois and Lyles, 1982) for 1 h at 37°C.
Subsequently, fixed cell sections were washed three times with 15 min incubation at 37°C in 3% Blotto + 0.5% Tween-20. A secondary antibody staining for 1 h at 37°C was then conducted using a combination of both 1:500 goat anti-rabbit immunoglobulin G (IgG)-Alexa 546 (Invitrogen, Carlsbad, CA) and 1:200 phalloidin-Alexa 488 (Invitrogen) or 1:500 goat anti-mouse IgG-Alexa 546 (Invitrogen) and 1:200 phalloidin-Alexa 488—depending on the species of primary antibody. Fixed cell sections were again washed three times with 15 min incubation at 37°C in 3% Blotto + 0.5% Tween-20, and nuclei were stained with 10 μM Draq5 (Biostatus Limited, Leicestershire, United Kingdom) + PBS for 20 min at room temperature, followed by three final washes in PBS. Stained cell sections were then mounted on microscope slides using medium set Vecta Shield (Vector Laboratories, Burlingame, CA), covered with number 1.5 cover slips (Fisher, Waltham, MA), and viewed using a Leica (Bannockburn, IL) SP5 confocal microscope either by imaging cell sections independently at 400 × (Airy 1.0, scan speed 400 Hz, sequential scan) or by z-plane stack imaging (90–100 slices per stack) through the entire cells at 630 × (Airy 1.0, scan speed 400 Hz, sequential scan). All images were captured at the same laser power settings.
VSV infections of neuroblastoma and nonneuronal cells for ultrastructural analysis
NB41A3 and L929 cells were plated into T175 flasks at 7 × 106 cells per flask and allowed to adhere to the growth substrate overnight. Afterward, cells were either treated with 400 U/mL IFNβ or medium alone for additional 18–24 h before infection. VSV was always added to cells in serum-free media, at a multiplicity of infection equal to 3, and allowed to adsorb for 30 min at 37°C, 5% CO2. Subsequently, unbound virus was removed by washing the monolayer several times with sterile Hank's balanced salt solution (Mediatech), and flasks were re-fed with complete media for the duration of the infection.
Embedding cellular samples for electron microscopy
After 6 or 7 hpi, supernatants were harvested for verification of VSV infection and IFNβ efficacy by plaque assay. The remaining cells were then washed one time in PBS and fixed in 3% paraformaldehyde + 0.5% glutaraldehyde + PBS for 30 min at room temperature. Cells were washed with PBS twice more and any remaining fixative was neutralized by 15 min incubation with a 1 M glycine (Sigma-Aldrich) + PBS solution at room temperature.
After one last wash with PBS, cells were dehydrated using increasing concentrations of ethanol under constant agitation as follows: 50%, 60%, 70%, and 80% ethanol washes for 30 min each, and 90%, 95%, and 100% ethanol washes for 1 h each. After dehydration, cells were treated with 5 mL amyl acetate for 1–2 min with repeated agitation to remove cell monolayers as sheets. Cell monolayers were then transferred into 50 mL conical tubes (Fisher) and washed with excess absolute ethanol to remove residual amyl acetate.
Subsequent to their dissociation as monolayers, cells were gradually infiltrated with increasing concentrations of LR White cut with absolute ethanol as follows: 3:1, 2:1, and 1:1 ethanol/LR White washes for 30 min; 1:2 and 1:3 ethanol/LR White washes for 1 h; and 100% LR White for greater than 1 h. Infiltrated cells were then transferred to BEEM® capsules (Electron Microscopy Sciences) and centrifuged at 1000 RCF for 10 min to pellet the cells toward the bottom of the capsule. Cells were then embedded within LR White by exposing the cell plus LR White to ultraviolet light for 48 h at 4°C. The embedded samples were cut into 50 nm sections using a Sorvall MT-2 ultra-microtome, and placed onto 300 mesh, thin bar nickel grids that had been previously washed with acetone.
Postembedded staining of cellular samples
Samples were blocked with 3% bovine serum albumin + 0.5% Tween-20 + 100 mM ammonium acetate for 30 min at room temperature by floating the grids containing the samples face down on 50 μL drops. Afterward, samples were probed with 1:100 rabbit anti-VSV P antiserum (a gift from Gail Wertz) (Green et al., 2000), 1:100 rabbit anti-VSV N antiserum (a gift from Gail Wertz) (Green et al., 2000), 20 μg/mL mouse anti-VSV M 23H12 monoclonal antibody (a gift from Doug Lyles) (Lefrancois and Lyles, 1982), or no primary antibody for 1 h at 37°C. Samples were subsequently washed three times by submerging each grid into consecutive 50 mL conical tubes containing ddH2O, followed by air-drying. Depending on the particular source of primary antibody used, samples were then treated with 1:40 goat anti-rabbit IgG conjugated with 6 nm gold particles (IgG-Au; Electron Microscopy Sciences), 1:40 goat anti-mouse IgG conjugated with 6 nm gold particles (Electron Microscopy Sciences), or no secondary antibody for 1 h at 37°C. Samples were again washed in ddH2O as described above and then fixed once more with 3% paraformaldehyde + 0.5% glutaraldehyde + PBS 30 min at room temperature.
After another round of washing, the samples were briefly floated on 100 mM cacodylate buffer and stained with 1% OsO4 in 100 mM cacodylate buffer pH 7.2 at room temperature for 30 min. Samples were washed in ddH2O as described above and then briefly floated on 100 mM ammonium acetate and stained with 4% uranyl acetate for 1 h at room temperature. Lastly, samples were subsequently washed three times by submerging each grid into consecutive 50 mL conical tubes containing ddH2O, followed by wicking off excess wash using a Kimwipe (Fisher) and air-drying. The grids were then ready for inspection and data were collected using a JEOL (Tokyo, Japan) 1200 XL transmission electron microscope.
Preparing virion samples for electron microscopy
To maximize viral titers, VSV infections of untreated NB41A3 or L929 cells had their supernatants harvested at 10 hpi, whereas supernatants from infections of IFNβ-treated cells were harvested at 18–24 hpi. In addition to the added time postinfection, the supernatants from four replicate T175 flasks containing untreated VSV-infected L929 or NB41A3 cells or eight T175 flasks with IFNβ-treated cells were pooled for each treatment group to increase viral particle recovery. Supernatants were then centrifuged at 1750 RCF and 4°C for 10 min to remove cell debris. Afterward, supernatants were carefully isolated and chilled on ice for 60 min, followed by additional centrifugation at ∼27,500 RCF and 4°C for 100 min to pellet virions.
Immediately after centrifugation, the supernatants were decanted and residual medium aspirated away, followed by the addition of 500 μL of ice-cold PBS + 0.53 mM EDTA. Viral pellets were allowed to sit in the PBS + 0.53 mM EDTA solution on ice for 1 h to loosen the pellets and were resuspended via pipetting and sonication. Resuspended crude VSV particles were further purified via ultracentrifugation at 42,000 RCF and 4°C for 90 min on a 5–40% sucrose gradient, where purified VSV migrated to the lower third of the gradient. The band of purified VSV was then isolated using a 1 cc tuberculin syringe (Becton Dickinson) and a 255/8G needle (Becton Dickinson); care was taken to minimize the volume isolated.
After purification, viral particles were fixed by mixing purified VSV with a 2 × fixative solution (6% paraformaldehyde + 1% glutaraldehyde + PBS) in a 1:1 ratio, followed by 30 min incubation at room temperature. Afterward, the fixatives were mixed with a 2 × neutralizing buffer (2 M glycine + PBS) at a 1:1 ratio; samples were incubated for an additional 30 min at room temperature. Fixed viral particles were then sprayed onto Formvar-coated 300 mesh nickel grids (Electron Microscopy Sciences) using a glass nebulizer. Samples were subsequently stained with 1% OsO4 in 100 mM cacodylate buffer pH 7.2 and 4% uranyl acetate as described above. The grids were then ready for inspection and data were collected using a JEOL 1200 XL transmission electron microscope. Viral particle length and width measurements were conducted at 100,000 × magnification using an Advanced Microscopy Techniques (AMT, Danvers, MA) Camera System in conjunction with AMT Advantage Software.
Computational analysis
Confocal images of immunofluorescently stained cell sections were analyzed using the Leica AF software package throughout the z-plane both independently and collectively—by collapsing all sections into a composite image. Stacked images were also exported from Leica AF as .avi movie files to better ascertain cellular localization patterns for VSV M, P, and N proteins during each time point postinfection. Stacked images were also projected, using the Volocity software package (Improvision, Coventry, United Kingdom), into three dimensions to further aid in observing localization patterns. All observations were compared across three independent experiments and were representative of those replicates.
Statistical analysis
For ultrastructural analyses, all experimental samples were prepared in triplicate, in at least three separate experiments. To determine the levels of budding virions from each cell type, the length of plasma membrane in electron micrographs at 75,000 × magnification was measured and the number of budding virions was counted across those membranes. The number of budding virions was then divided by the length of plasma membrane and expressed as budding particles per micron ± 95% confidence intervals. For assessing the size of viroplasmic regions in the various treatment groups, the number of 6 nm gold particles was counted across a 75,000 × viewable field and represented as an average ± the standard error of the mean. For calculating the statistical relevance for viral particle size, sample t-values were calculated using Satterthwaite's method for independent samples of unequal variances, and hypothesis testing was employed to determine whether or not viral particle dimensions were equal, yielding p-values indicative of these tests. The 95% confidence intervals for each mean were also calculated.
Results
The progression of VSV infection in fibroblasts is completely abrogated by a type I IFN response
VSV replication is quite sensitive to IFN treatment; cells treated with IFNβ yield three to four logs lower infectious VSV titers (Fig. 1). However, despite its impact on VSV titers, type I IFN treatment of neuroblastoma cells elicits a different response than similarly treated fibroblasts (D'Agostino et al., 2009). VSV proteins are undetectable in lysates from IFNβ-treated fibroblasts, whereas VSV proteins are still found in IFNβ-treated neuroblastomas—although at 3–12-fold lower levels than control cells (Trottier et al., 2005; D'Agostino et al., 2009). Further experimental evidence has demonstrated IFNβ-induced changes in the posttranslational modifications of VSV M and P proteins from neuroblastoma cells (D'Agostino et al., 2009). These observations have led to the hypothesis that a nontraditional type I IFN response is acting to disrupt the VSV infectious cycle at the assembly/budding stage.
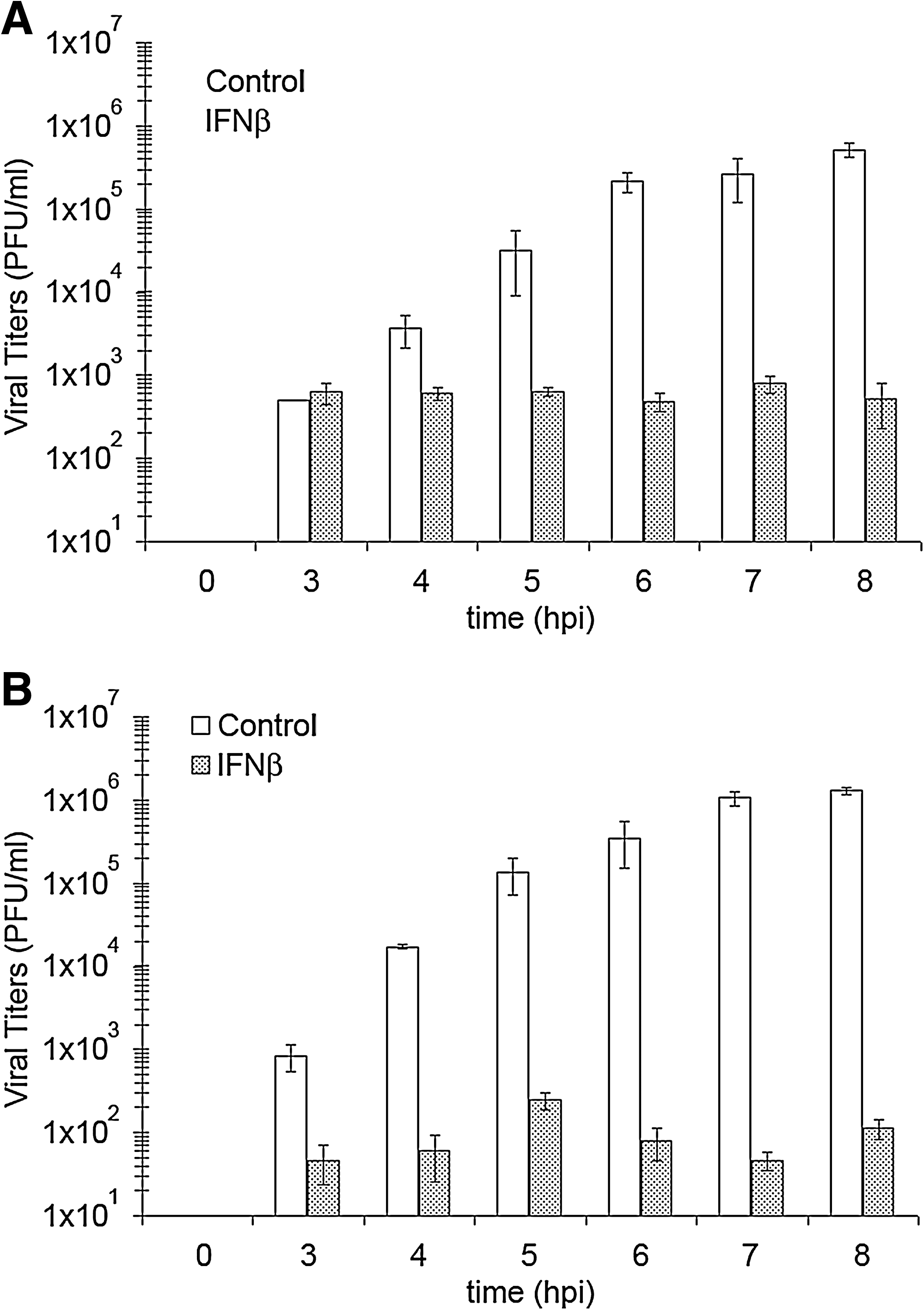
Infectious VSV particles released from neuronal and nonneuronal cells during an 8 h time course. Supernatants were harvested from VSV-infected (multiplicity of infection = 3) (
To test our hypothesis, we first set out to study VSV infections of the fibroblast/adipocyte-derived L929 cell line. VSV aggressively spreads through most cell lines in vitro within 8 hpi. At 8 hpi, when examined by phase contrast microscopy, greater than 95% of cells showed cytopathic effects (CPEs), as defined by rounding in cell morphology, and after that point cells began to detach from their growth substrate and float in suspension (data not shown). Using immunofluorescence confocal microscopy, we tracked VSV infections of L929 cells treated with 400 U/mL IFNβ or vehicle alone by staining for three VSV proteins: VSV M, P, and N.
Over an 8 h time course viral titers steadily increased up to ∼5 × 105 PFU/mL in untreated cells, whereas VSV titers remained just above the limit of detection for plaque assays in IFNβ-treated L929 cells (Fig. 1A). In line with the observed increases in VSV titers, expression of VSV M (Fig. 2A) in L929 cells increased over time and hit a maximum expression level at 6 hpi (data not shown). The VSV M protein staining pattern appeared both cytoplasmic and nuclear throughout the time course. The staining patterns for VSV P and N proteins showed similar expression profiles as demonstrated by only 7 hpi for each staining (Fig. 2B, C). However, while VSV P appeared mostly cytoplasmic with some punctate expression patterns during the entire time course, VSV N staining started out cytoplasmic and by 5 hpi appeared more structured into larger asymmetrically stained foci (data not shown). Interestingly, at 7 and 8 hpi, CPEs were present in a majority of L929 cells, but only half of those cells were expressing detectable levels of VSV M, P, and N proteins.

VSV progression through nonneuronal L929 cells during an 8 h time course and the IFNβ effect on that progression. VSV protein expression was seen steadily increasing in greater numbers throughout the course of infection in untreated L929 cells by observing cells stained with (
VSV infections of IFNβ-treated L929 cells showed no VSV M protein expression throughout the infection time course (Fig. 2D). Similarly, no expression of VSV P or N proteins was found in IFNβ-treated L929 cells (data not shown). These results were in agreement with previous observations showing undetectable expression of VSV proteins in IFNβ-treated infected L929 lysates (D'Agostino et al., 2009).
The progression of VSV infection in neuroblastomas is attenuated by an IFNβ response
Next we followed VSV infections of NB41A3 neuroblastomas, treated with 400 U/mL IFNβ or vehicle alone, by both plaque assay and by immunofluorescence confocal microscopy of VSV M, P, and N proteins. Similar to the L929 VSV time course infections, we observed a steady increase in viral titers from untreated NB41A3 cells—plateauing ∼1 ×106 PFU/mL—and barely detectable titers from IFNβ-treated cells (Fig. 1B). VSV protein expression within NB41A3 cells matched viral particle production, as we saw a steady increase in cells positively stained for the VSV M (Fig. 3A), P (Fig. 3C, left panel), and N (Fig. 3D, left panel) proteins. In addition, the intracellular staining patterns observed for each viral protein were indistinguishable from those seen in L929 experiments (Fig. 2).
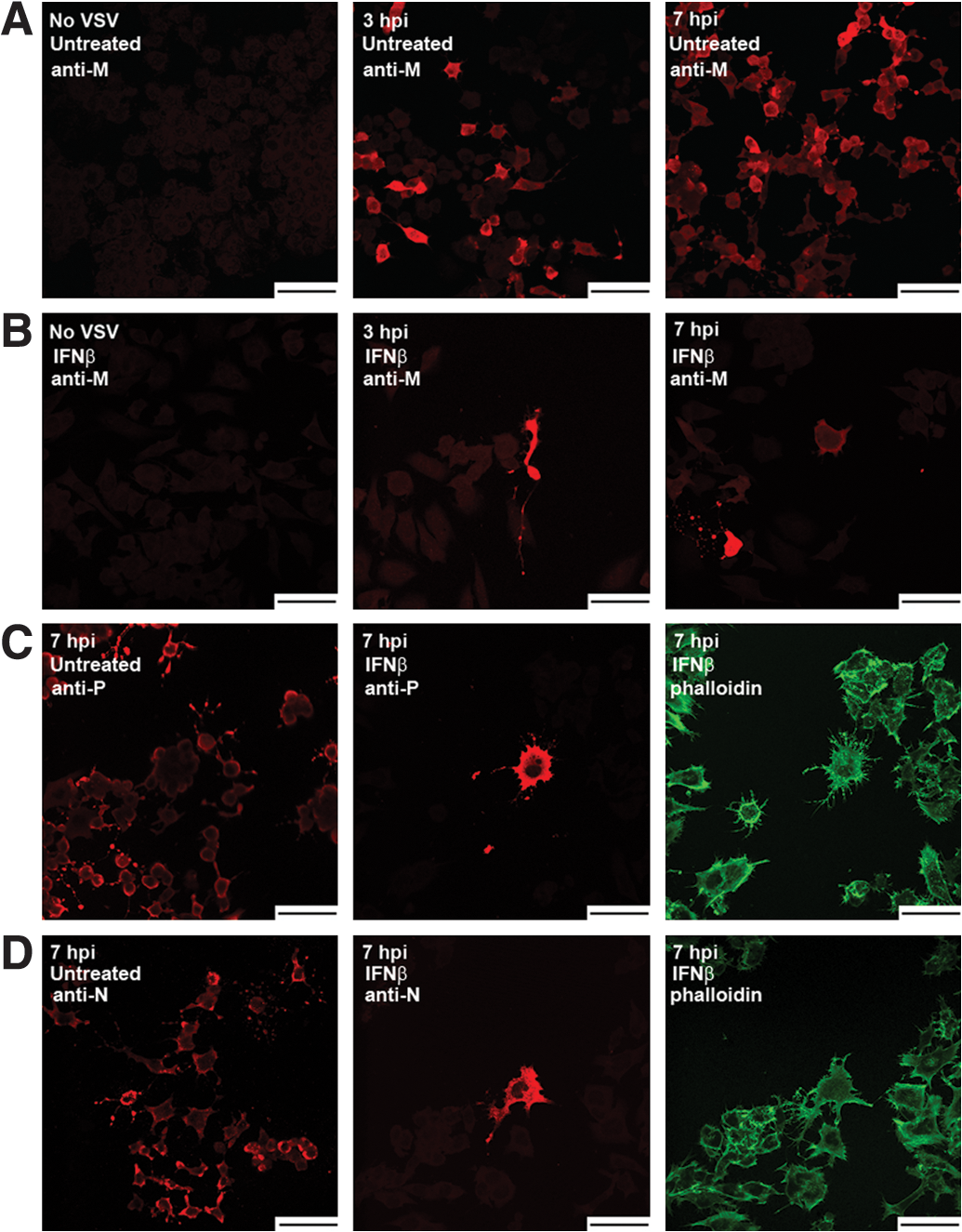
VSV viral protein expression patterns in IFNβ-induced NB41A3 neuroblastoma varies greatly from that in similarly treated L929 cells. VSV-infected, (
Unlike L929 cells, IFNβ-treated, VSV-infected NB41A3 cells appeared to change in morphology, as these cells appeared to become more spread out along the growth substrate (data not shown). In contrast to L929 cells, IFNβ-pretreated NB41A3 cells did show expression of VSV proteins. This observation was expected from our earlier experiments in which Western blots were performed on infected cell lysates (Trottier et al., 2005; D'Agostino et al., 2009). Unexpectedly, however, there was a small fraction of IFNβ-treated neuroblastoma cells, on the order of 1-out-of-10,000, at 3 hpi that showed immunofluorescence for VSV M (Fig. 3B), P (Fig. 3C, middle and right panels), and N (Fig. 3D, middle and right panels) proteins. The number of positively stained IFN-treated NB41A3 cells then slightly increased by one to two per 104 cells every hour for the duration of the experiments with positive cells always appearing in small groups, suggesting a slowed spread of the virus. When these IFN-treated cells were allowed to incubate for 16–24 h, the infection would progress through all the cells, leaving advance-staged CPEs and dead cells floating in suspension (data not shown). In contrast, IFNβ-treated, VSV-infected L929 cells remained apparently healthy for greater than 24 hpi, until cell density became an issue. This indicated that IFN treatment of L929 cells completely suppressed viral replication but was not cytostatic. These data were consistent with previous data that showed a modest decrease in both VSV transcript and viral protein expression within IFNβ-treated infected NB41A3 lysates (Trottier et al., 2005). We interpreted these observations as attenuation in VSV virulence resulting from an IFNβ-induced antiviral state.
Intracellular distribution of VSV proteins in IFNβ-treated neuroblastomas is not different from that in L929 cells despite attenuation of viral replication
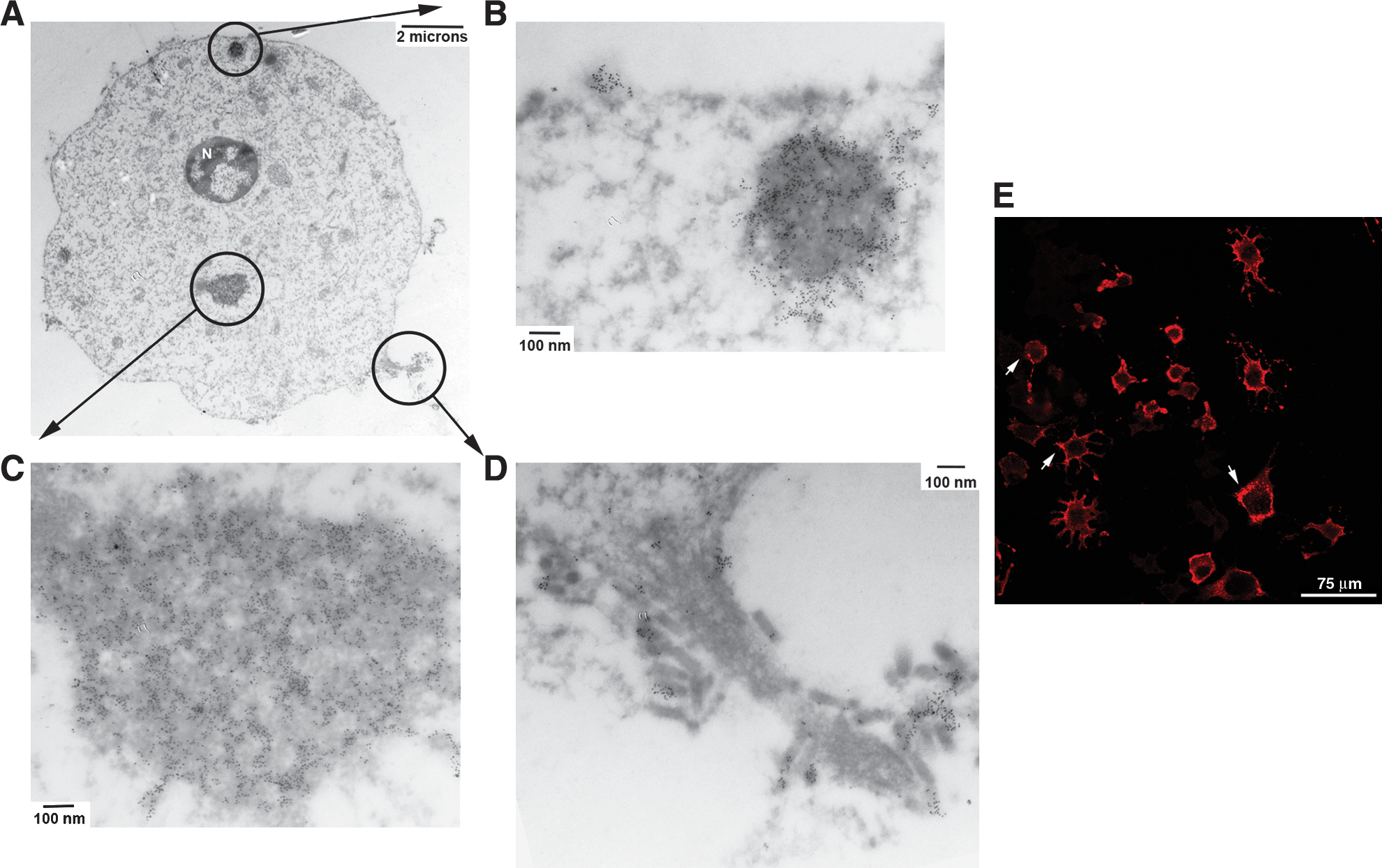
Given the attenuated nature of VSV infections in IFNβ-treated neuroblastomas, we examined whether differences in the intracellular localization of the various viral proteins might explain the observations. Stacked confocal images were analyzed at a higher magnification for immunofluorescent staining of VSV M, P, and N proteins. Typically, cells were 10–13 μm thick and 90–100 slices were performed per stack. Stacked images were viewed as independent slices, exported as movies, and projected three-dimensionally to identify any patterns associated with VSV protein localization.
The intracellular distribution pattern associated with the VSV M protein found in untreated NB41A3 cells was consistent with both cytoplasmic and nuclear localizations. Progression through the various stacks identified M staining patterns that overlapped with the Draq5 nuclear marker, as well as staining that filled out the cytoplasmic boundaries highlighted by cytoskeletal staining with phalloidin (Fig. 4A, left panel). This staining pattern was supported by three-dimensional projections, which clearly showed staining of the entire neuroblastoma cell body and nuclear envelope (data not shown). A similar analysis of VSV M distribution in IFNβ-treated NB41A3 cells showed no significant deviations from those patterns identified in untreated cells (Fig. 4A, right panel).

The VSV protein localization in IFNβ-induced NB41A3 neuroblastomas is no different from its distribution in untreated cells. (
Analysis of VSV P protein distribution in infected, untreated NB41A3 cells revealed mostly uniform cytoplasmic staining (Fig. 4B, left panel). Some cells also had an additional punctate localization pattern for VSV P; however, this was not always observed under these staining conditions. Compared with untreated cells, IFNβ-treated, VSV-infected NB41A3 cells had no significant difference in VSV P protein localization, which was also supported by three-dimensional projections (Fig. 4B, right panel, and 4D).
Lastly, we studied the staining patterns of the VSV N protein in both IFNβ- and control-treated neuroblastomas. Within untreated NB41A3 cells, we saw uneven cytoplasmic staining, with areas of heavily stained cytoplasm located near cell membranes (Fig. 4C, left panel). Like the other viral proteins, VSV N localization did not significantly change in IFNβ-treated, virally infected neuroblastomas (Fig. 4C, right panel). Overall, we were unable to attribute the observed IFNβ-induced attenuation in VSV virulence to any gross changes in viral protein distribution.
General ultrastructural observations of VSV-infected L929 and NB41A3 cells
The VSV infectious cycle has been shown to occur entirely within a host cell's cytoplasm (Rose and Whitt, 2001), which can be broken down into five stages: adsorption, entry/uncoating, replication, assembly, and budding. During latter stages of the VSV infectious cycle CPEs begin to arise, caused mostly by the M protein, with contributions from the G protein (Florkiewicz et al., 1983; Blondel et al., 1990; Melki et al., 1994; Ye et al., 1994; Lyles and McKenzie, 1997). This phenomenon holds true for VSV infections of L929 (Fig. 5A) and NB41A3 (Fig. 5C) cells. In both cells the CPE observed is the result of a disruption in cell morphology whereby the contacts each cell has with the growth substrate are mostly lost, leaving only a small foot tethered to the remaining cell body (as seen in the L929 sagittal view with relation to the extracellular matrix from Fig. 5A). When observed from above these infected cells experiencing CPEs take on a rounded appearance (as seen in the transverse view for an NB41A3 cell from Fig. 5C). In addition to a rounded morphology, cells experiencing CPEs also displayed possible signs of apoptosis that included bloated nuclei, less densely stained cytoplasm, and compromised cell membranes. However, L929 cells pretreated with IFNβ maintained a normal appearance despite being exposed to infectious virus (Fig. 5B). For the most part the same was true of IFNβ-treated NB41A3 cells infected with VSV, except for the low frequency of cells displaying CPEs, which were on the order of those cells expressing viral proteins observed via confocal microscopy (Fig. 5D).
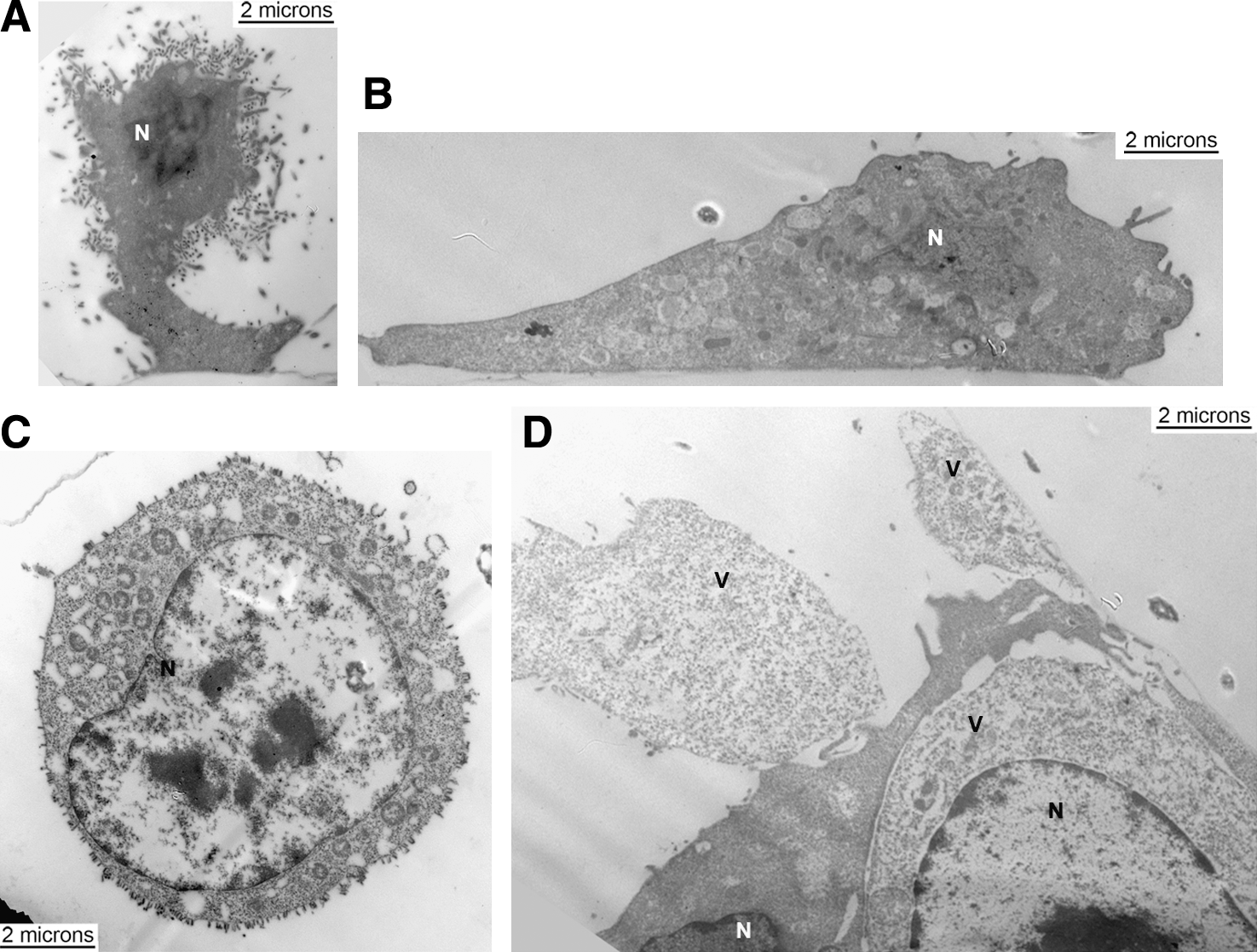
General observations on VSV-infected L929 and NB41A3 cells with and without IFNβ treatment at 18–24 hpi. (
IFNβ-treated, VSV-infected L929 cells show no signs of viral infection
Upon closer examination of VSV-infected L929 cells, by 6 hpi the entire plasma membrane surface is marked with budding virions (Fig. 6). Additionally, after staining cell sections with rabbit anti-VSV P antiserum and goat anti-rabbit IgG-Au, the presence of viroplasmic regions became evident and could possibly correlate with aggregate anti-N staining typically viewed via confocal microscopy (Fig. 6). These viroplasmic regions were electron dense and contained concentrated VSV P proteins. VSV N protein (Fig. 7A) and VSV P protein (Fig. 7B) were observed in viroplasm close to membranes with actively budding virions. However, L929 cells pretreated with IFβ displayed neither electron-dense structures nor staining for either VSV P or N proteins (Fig. 8). This is consistent with the biochemical data (D'Agostino et al., 2009).
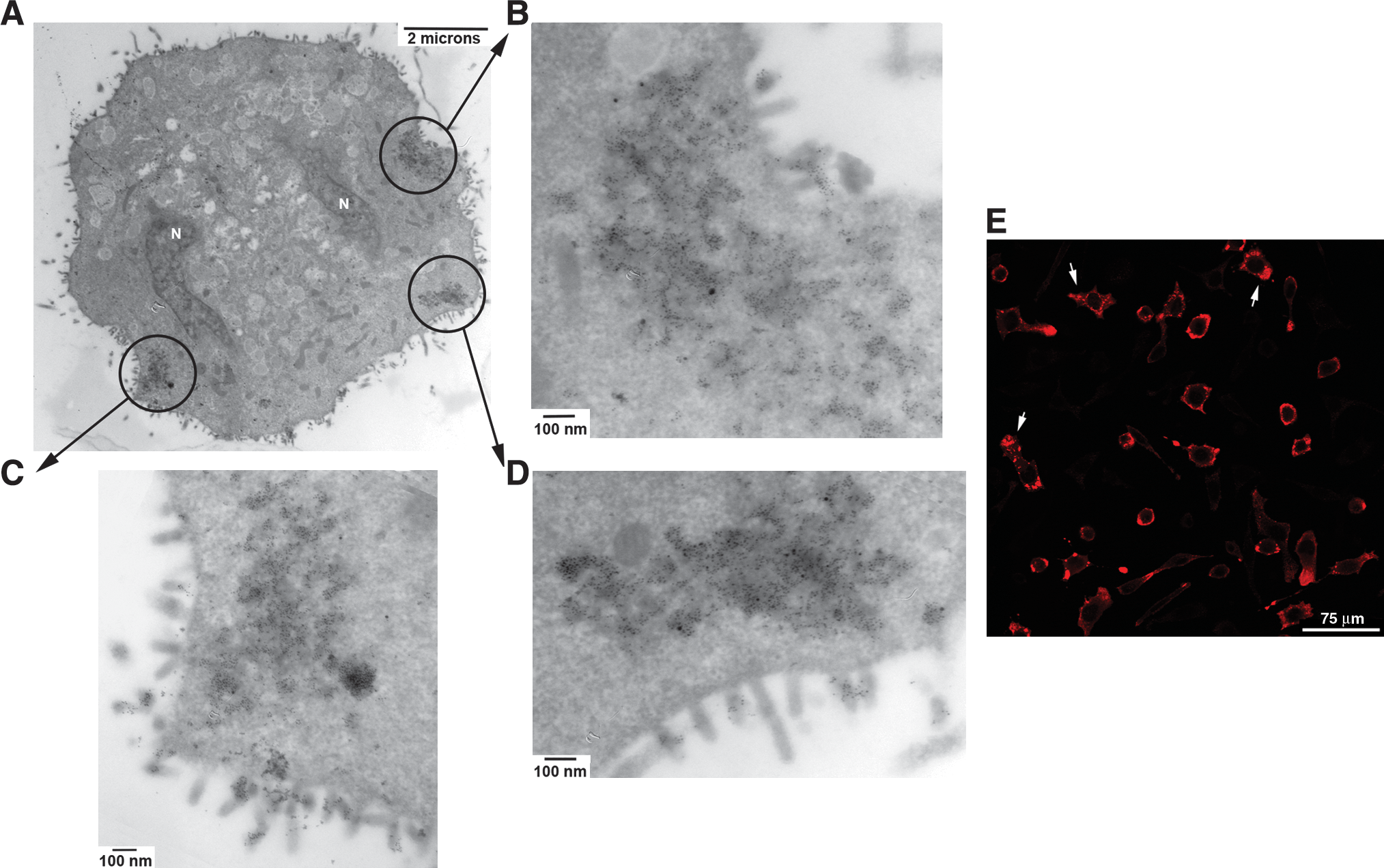
Untreated, VSV-infected L929 cells have electron-dense regions containing viral proteins at 6 hpi. (
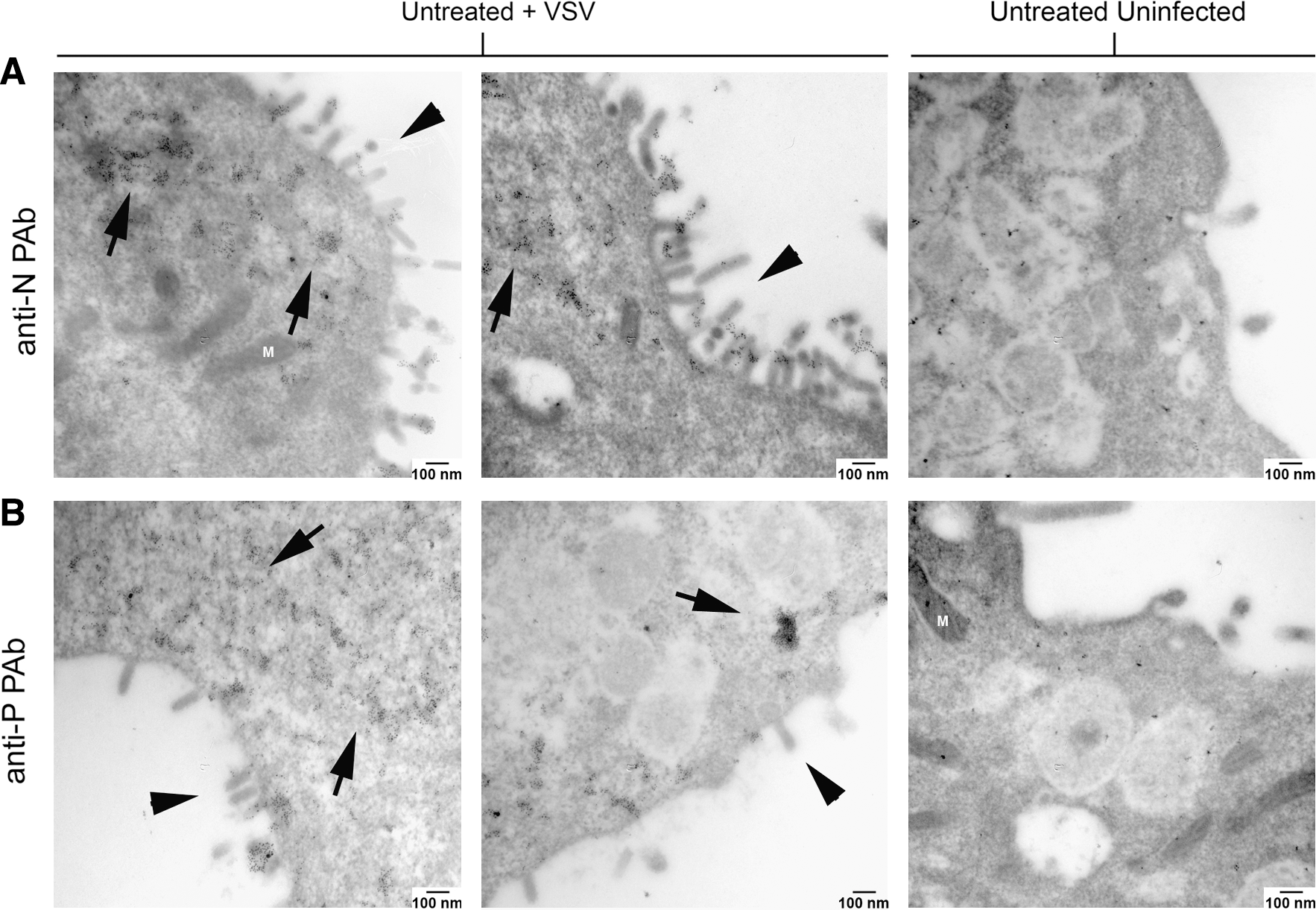
Viroplasm-like regions within untreated, VSV-infected L929 cells contain both VSV P and N proteins. (
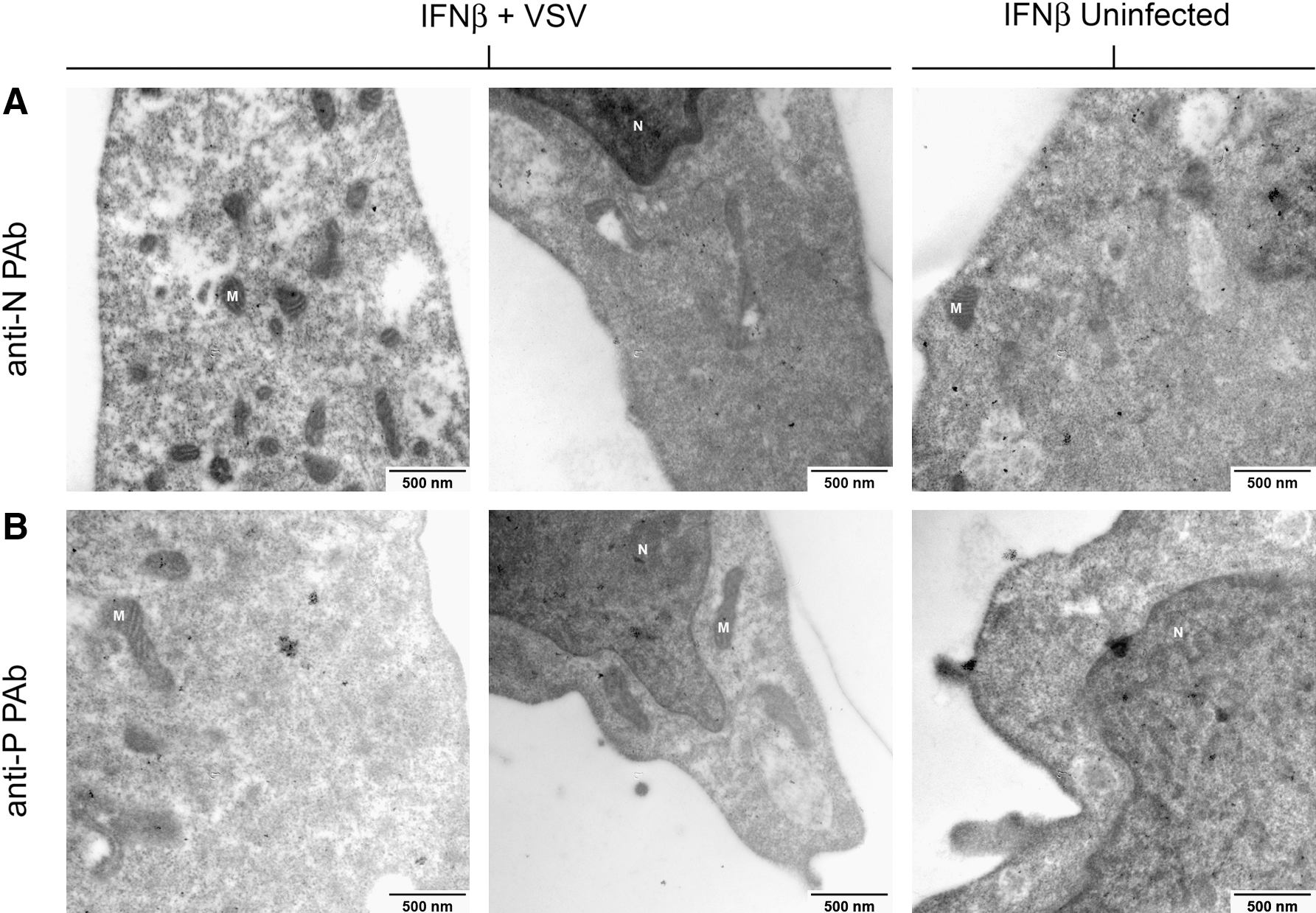
Viroplasmic regions are not found in IFNβ-treated, VSV-infected L929 cells at 6 hpi. IFNβ-treated, VSV-infected L929 cells at 6 hpi and IFNβ-treated, uninfected L929 cells were probed in a similar fashion as the cells in Figure 7. After examining 20–30 electron micrographs per treatment group, at 75,000 × magnification, neither electron-dense regions nor the expression of (
Similar experiments were conducted by processing embedded samples with the monoclonal anti-VSV M antibody (23H12), but resulted in no detectable staining even in control samples. We interpret these data to indicate that the epitope recognized by 23H12 was destroyed by the embedding, fixing, or staining procedures.
IFNβ-treated, VSV-infected NB41A3 cells showed signs of an attenuated viral infection
VSV infections of untreated NB41A3 neuroblastomas also resulted in electron-dense regions, although these regions did not always occur near membranes where VSV budding was taking place (Fig. 9). Again, these viroplasmic regions seem to corroborate aggregate anti-N staining viewed during our confocal microscopy studies. Another apparent difference between VSV-infected untreated NB41A3 and L929 cells, was that overall numbers of budding virions from L929 cells (Figs. 5A and 6A) were greater (10.9 ± 2.6 virions per micron of plasma membrane) than those budding from NB41A3 cells (Figs. 5C and 9A; 4.8 ± 1.8 virions per micron of plasma membrane, p = 0.0006). This was not surprising since we have anecdotally observed disproportionate levels of viral production with VSV infections of different cell types, for example, BHK-21, CHO, NIH/3T3, and L929 (data not shown). Viroplasmic regions in NB41A3 cells were generally smaller in scale and contained lower levels of staining for VSV N (Fig. 10A; on average 174 ± 41 gold particles per 75,000 × viewing field) and P proteins (Fig. 10B; 264 ± 98 gold particles per 75,000 × viewing field) than those observed in L929 cells (237 ± 53 gold particles per 75,000 × viewing field for anti-N staining and 469 ± 144 gold particles per 75,000 × viewing field for anti-P staining).
Untreated, VSV-infected NB41A3 cells show viroplasm-like regions containing viral proteins at 6 hpi. (
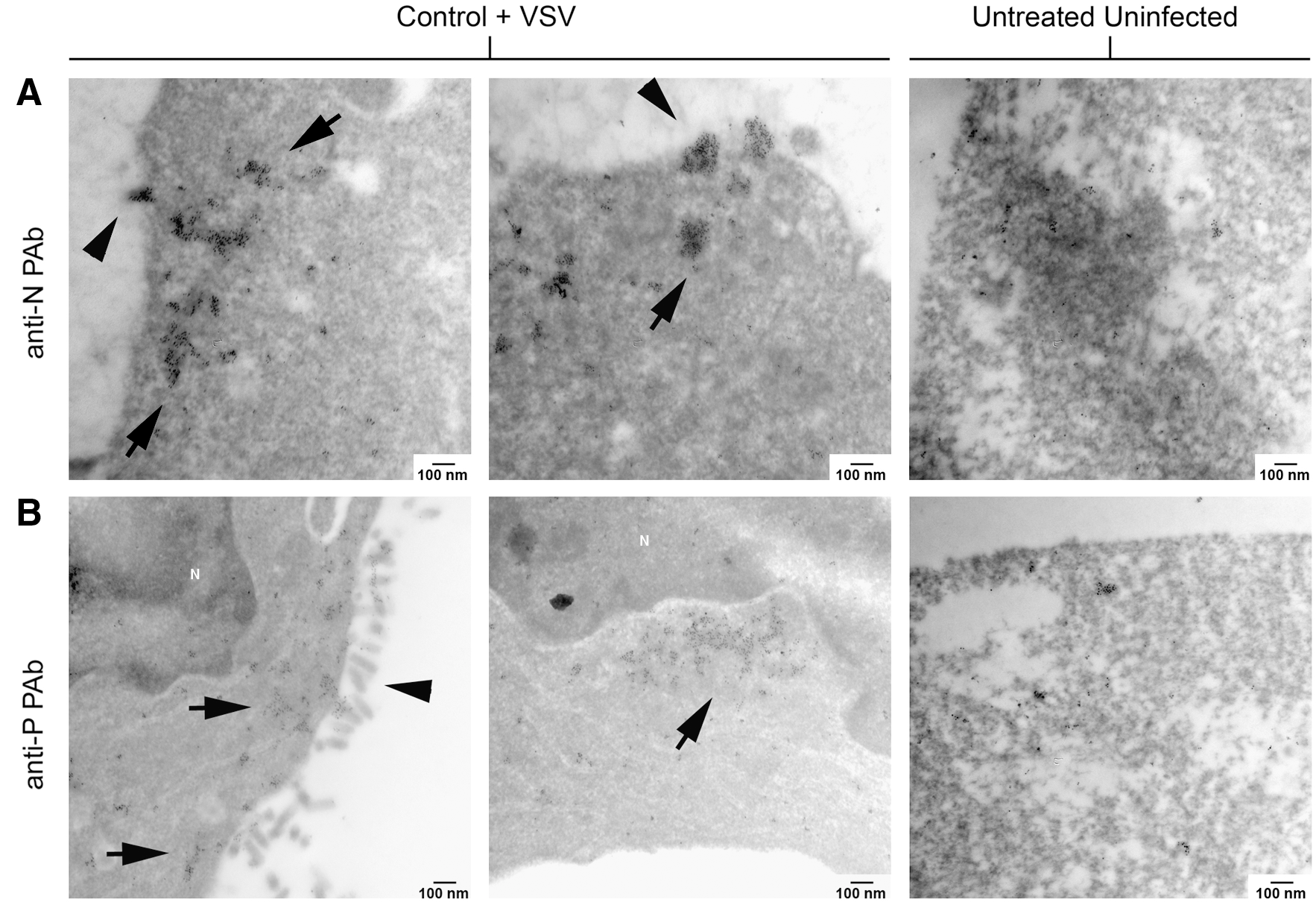
Viroplasmic regions within untreated, VSV-infected NB41A3 cells also contain both VSV P and N proteins. Untreated, uninfected or untreated, VSV-infected NB41A3 cells were stained as described in Figure 7. Electron micrographs at 75,000 × magnification show viroplasm-like regions colocalized with VSV N and P proteins, as observed by the collection of 6 nm gold particles. Unlike L929 cells, these viroplasmic regions were not near regions of active virion budding and were not as heavily electron dense. (
Unlike L929 cells, IFNβ-treated NB41A3 cells did show staining for VSV N and P proteins (Fig. 11). NB41A3 cells positively stained for viral proteins, however, were difficult to find. When positive cells were found, they were comparable to the number of cells expressing viral proteins observed via confocal microscopy. In addition to their infrequency, viroplasmic regions in IFNβ-treated NB41A3 cells were noticeably smaller (103 ± 9 gold particles per 75,000 × viewing field for anti-N staining and 82 ± 28 for anti-P staining) and not as electron dense as those observed in untreated neuroblastomas; in some cases these regions were difficult to separate from background staining (Fig. 11A). Lastly, the viroplasm found in IFNβ-treated, VSV-infected NB41A3 cells was not located near actively budding virions. In fact, we were unable to identify IFNβ-treated, VSV-infected NB41A3 cells budding any viral particles at 6 hpi.

Unlike L929 cells, small viroplasmic regions are found in IFNβ-treated, VSV-infected NB41A3 cells and contain both VSV P and N proteins at 6 hpi. IFNβ-treated, VSV-infected NB41A3 cells at 6 hpi or IFNβ-treated uninfected were again stained as in Figure 7. Electron micrographs at 75,000 × magnification show substantially smaller electron dense regions colocalized with either (
Ultrastructural differences between VSV virions harvested from infected L929 and NB41A3 cells and IFNβ effects on those differences
Next, we turned our attention to the viral particles released from VSV-infected L929 and NB41A3 cells. VSV virions harvested from untreated L929 cells had the bullet-shape morphology characteristic of the Rhabdoviridae family with an average length of 146.5 ± 3.9 nm and a two-dimensional projected width of 51.5 ± 1.6 nm (Fig. 12A). Viral particles examined from untreated, VSV-infected NB41A3 cells also appeared bullet shaped and had on average a 151.0 ± 4.9 nm length and a 46.7 ± 2.2 nm width (Fig. 12B).

VSV viral particles that budded from IFNβ-treated NB41A3 cells contained characteristic bullet shape as well as pleomorphic virion-like particles. Supernatants from VSV infections of untreated NB41A3 or L929 cells were harvested at 10 hpi and supernatants from IFNβ-treated cells were harvested at 18–24 hpi. Viral particles were purified and then fixed then stained with 1% osmium tetroxide in 100 mM cacodylate buffer pH 7.2 and 4% uranyl acetate. Electron micrographs at 100,000 × of VSV viral particles from (
To assess ultrastructural changes that may have resulted from IFNβ, we increased the number of infected flasks, extend the time postinfection until harvesting viral supernatants, and include samples from crudely purified virus. Despite these modifications to our recovery protocol, we were unable to identify VSV virions from IFNβ-induced L929 cells. However, we were able to detect viral particles from IFNβ-treated, VSV-infected NB41A3 cells with a bullet shapes and dimensions of 138.1 ± 11.0 nm length and a 49.7 ± 9.3 nm width although from a very small sample size (n = 5; Fig. 12C). Statistical analysis to determine the observed dimensional differences between virions from untreated and IFNβ-treated NB41A3 cells revealed no difference among treatment groups. Similar tests conducted between untreated L929 and NB41A3 cells revealed no statistical difference among cell types. We interpret these data to indicate that the virions were morphologically indistinguishable.
Another population of virus-like particles was observed in the neuroblastoma IFNβ-treated group, which was not present in the untreated NB41A3 or L929 samples. In addition to the characteristic bullet morphology with typical dimensions, more elongated particles were identified as well as peculiar shapes, for example, spherical and conical structures (Fig. 12D). However, due to the sticky nature of the viral particles and the very low number of recovered virions, there was a high degree of uncertainty in identifying whether these IFNβ-induced aberrant morphologies had VSV antigens.
Discussion
Given the unique immunological environment represented by the CNS and the large number of ISGs, it should not be surprising that the antiviral mechanisms in neuroblastoma cells differ from those in nonneuronal cell lines. Previous evidence has demonstrated that VSV infections of IFNβ-treated fibroblasts and neuroblastomas resulted in a substantial decrease in infectious viral titers when compared with similar infections of untreated cells (D'Agostino et al., 2009).
While this was not surprising, the mechanism that IFNβ elicited to curtail VSV replication was unexpected. Examination of fibroblast-derived cellular lysates by either Western blot or immunofluorescence microscopy revealed undetectable levels of VSV proteins. Such evidence was consistent with the well-elucidated pathways that act to inhibit viral infection by shutting down host protein production, either by preventing the recycling of translation initiation factors or by degrading mRNA (Goodbourn et al., 2000). However, similar experiments done with neuroblastomas deviated from these well-studied pathways in that substantial levels of viral proteins and even viral transcripts were observed (Trottier et al., 2005; D'Agostino et al., 2009). In addition, biochemical analyses of the viral proteins from IFNβ-treated, VSV-infected neuroblastomas demonstrated hyperphosphorylation of the M protein, which resulted in a disruptions in its interactions with the N protein (D'Agostino et al., 2009). This evidence pointed to a possible defect in VSV assembly in neuroblastoma cells induced by type I IFN.
Using immunofluorescence confocal microscopy, we tracked VSV M, P, and N proteins during an 8 h infection of neuroblastoma and fibroblast cell lines. We chose an 8 h infection since that was the time it took for the vast majority of untreated cells to exhibit CPEs while still remaining bound to the growth substrate. During the infections, cells were frozen in time via chemical fixation at every hour starting at 3 hpi.
VSV replication progressed similarly in untreated NB41A3 neuroblastoma and L929 fibroblast cell lines (Fig. 1). Infection in both cell lines showed an increasing number of VSV-positive cells over time, with a maximum frequency at ∼6 hpi (Figs. 2A–C and 3A). The intracellular distribution patterns for each viral protein agreed with expectations, based upon each proteins function during the VSV infectious cycle. VSV M protein was always found localized to both nuclei and cytoplasm (Fig. 4A). VSV M protein cytoplasmic staining appeared uniform without significant changes in protein distribution throughout the observed time course. The intracellular localization pattern of VSV M agreed with its function as an inhibitor of host mRNA export, mitochondrial binding, and its role in virion assembly (Lyles et al., 1988; Jayakar et al., 2004; Lichty et al., 2006).
The localization pattern for VSV P protein was strictly cytoplasmic in both neuroblastomas and fibroblasts. However, unlike VSV M protein, VSV P protein was detected in both punctate and diffuse staining patterns, which remained for the duration of the time course (Fig. 4B). The punctate staining could be representative of viroplasm—high-density areas of newly synthesized viral products—which are in line with VSV P function in viral genome replication and transcription. Likewise, the distribution of VSV N protein was also cytoplasmic with high-density staining foci in both NB41A3 and L929 cells (Fig. 4C). Additionally, the VSV N-associated high-density areas were larger and more asymmetrically distributed than those observed when staining for VSV P protein. Another deviation from VSV M and P proteins was that VSV N distribution changed during the infectious time course from relatively diffuse staining early to higher density patterns later (data not shown). This distribution is consistent with free and RNA-bound VSV N, as well as its early roles in viral RNA protection and RDRP interactions; the later patterns are consistent with its role during assembly (Chong and Rose, 1994; Barr et al., 2002; Jayakar et al., 2004; Gerard et al., 2007). Similar distribution patterns for VSV P and N proteins have also been reported in BHK-21 cells under live cell microscopy (Das et al., 2006).
The similarities between VSV infections of NB41A3 and L929 cell lines stopped when those cells were exposed to IFNβ before infection. In L929 fibroblast-derived cells, IFNβ treatment resulted in undetectable expression of VSV M, P, and N proteins during the 8 h observation period (Fig. 2D). In fact, no VSV protein expression was detected even at 24 hpi (data not shown). However, with IFNβ pretreatment of NB41A3 cells, VSV infection led to viral protein expression for VSV M, P, and N as early as 3 hpi, when we observed brightly positive cells at a frequency of approximately 1 cell per 10,000. These lower numbers of brightly immunofluorescent cells did progress at a very slow rate through the neuroblastomas for the duration of the time course suggesting the spread of VSV (Fig. 3B–D). In contrast to L929 cells, VSV infection of IFNβ-treated NB41A3 cells persisted until all cells succumbed to the infections between 16 and 24 hpi (data not shown). These observations point to an IFNβ-induced attenuation in VSV virulence and offer explanation as to why only a 3–12-fold reduction in viral products is typically seen in VSV infections of type I IFN-treated neurons (Trottier et al., 2005).
To identify anomalies in VSV protein localization that may be responsible for the observed attenuation in IFNβ-treated neuroblastomas, immunofluorescence confocal microscopy was used to create stacked image composites of VSV-infected cells using 90–100 sections per cell. These images were viewed on a section-by-section basis, as a collapsed image of all sections, and by virtual three-dimensional projections. Such analyses of the VSV M protein from IFNβ-treated neuroblastomas revealed no differences in the protein's localization when compared with untreated cells for the duration of the time course (Fig. 4A). Subsequent analyses of VSV P and N protein yielded similar results; that is, each protein's localization pattern was analogous to its pattern in untreated cells (Fig. 4B–D).
Our observations have helped to explain some of the discrepancies between a type I IFN response in nonneuronal versus neuroblastoma cells. In nonneuronal cells (e.g., L929 fibroblasts) viral protein production is shut down by a cessation of host processes at the transcriptional and translation levels processes (Belkowski and Sen, 1987; Stojdl et al., 2000). We did not observe viral protein expression even at greater than 24 hpi of IFNβ-treated L929. Neuroblastoma cells, on the other hand, had been show to deviate in their response to IFNβ exposure, since modestly lower levels of viral transcripts and proteins are still detected in these cells when infected with VSV (Trottier et al., 2005). This lower level of viral products is due to attenuation, not abrogation, of VSV RNA and protein synthesis within IFNβ-treated neuroblastoma cells. Although VSV growth is attenuated—but still viable—in neuroblastoma cells treated with type I IFN, we still do not understand the mechanism(s) of this attenuation. We do know that the VSV M and P protein have altered phosphorylation within a neuronal type I IFN-induced antiviral state, and that hyperphosphorylation of VSV M disrupts its interactions with VSV N protein (D'Agostino et al., 2009). However, other important cellular pathways may also be involved in the assembly and budding defects we observed.
Interactions between the M protein and VSV RNP core, via N, are crucial in the assembly process. However, the confocal methods we employed in this study were unable to identify any changes in viral protein localization that may point to virion misassembly as the cause for VSV attenuation. We then shifted our attention to an ultrastructural examination VSV infections of IFNβ-treated neuroblastomas. Immediately, a difference in the extent of emerging viral progeny was identified between the nonneuronal (L929) and neuroblastoma (NB41A3) cell lines infected with VSV. L929 cells displayed a far greater number of budding VSV virions across its plasma membrane compared with NB41A3 cells (Fig. 5A, C). Despite the difference in budding particles, both L929 and NB41A3 cells showed signs of apoptosis after 6 to 7 hpi and possessed electron-dense, VSV protein containing regions (Figs. 6 and 9) similar to the viroplasmic regions found with other viruses and host cells (Netherton et al., 2007).
Several other notable differences between L929 and NB41A3 cells became apparent when VSV infections of IFNβ-treated cells were compared. VSV-infected, IFNβ-treated L929 cells showed no signs of infection when examining either the overall cell appearance (Fig. 5C) or when probing for VSV proteins using VSV-specific antibodies (Fig. 8). However, VSV-infected, IFNβ-treated NB41A3 cells did show signs of apoptosis (Fig. 5D) and expression of viral proteins (Fig. 11), but these occurred in a very small fraction of cells, were difficult to find when scanning embedded sections with the electron microscope, and indicated no signs of budding virions. In addition, the viroplasmic regions observed in untreated NB41A3 cells were also apparent in IFNβ-treated cells. However, those cytoplasmic viroplasmic regions in IFNβ-treated NB41A3 cells appeared significantly smaller and were sometimes difficult to differentiate from background staining.
Unfortunately, we were unable to identify gross changes within VSV-infected, IFNβ-treated cells that supported virion misassembly by using staining for viral proteins alone. Future experiments will require either dual staining with markers for microtubules, the multivesicular body, or other cellular structures relevant to vesicle partitioning. In addition, identification of ISGs in both neuroblastoma and nonneuronal cells by microarray could provide potential targets for RNAi to help pin-point possible host cell effectors whose actions either directly or indirectly effect virion assembly.
Finally, since we were unable to identify cellular indictors of virion misassembly, we chose to examine the ultrastructure of the VSV particles. Viral particles from untreated L929 or NB41A3 cells possessed the bullet shape characteristic of VSV (Fig. 12A, B) and the dimensions of these viral particles were shown to be statistically similar. As was expected, no viral particles were isolated and observed from IFNβ-treated L929 cells. However, with much effort and resource expenditure we were able to isolate and examine virions from IFNβ-treated NB41A3—although with a much smaller sample size than from untreated cells. The particles we could positively identify as VSV (i.e., possessing bullet shapes) were dimensionally indistinguishable to those from untreated NB41A3 and L929 cells (Fig. 12C).
Additional pleomorphic viral-like particles were also present in crude VSV preparations, but were either of odd dimensions or shapes from what we typically observed (Fig. 12D). However, we cannot determine with any certainty if these pleomorphic virus-like particles actually represent misassembled VSV due to their tendency to nonspecifically bind antibody and their very low numbers. These particles do give rise to speculation of either misassembly or an increase in defective particles resulting from an IFNβ-induced antiviral state in neuroblastoma cells. The fact that these particles were only observed in crude VSV isolates, generated through pelleting, and not in VSV purified through sucrose gradients will require modified processing similar to those methods used in identification of defective interfering particles (Huang et al., 1966; Holland and Villarreal, 1975).
Our experiments have demonstrated compelling evidence that type I IFN-elicited responses in neuroblastoma cells differ profoundly from those responses in the more traditionally used fibroblast-derived L929 cells. These observations are consistent with previously observed biochemical and viral growth differences between neuroblastoma and nonneuronal cells, and could point to a novel type I IFN-induced antiviral mechanism in neurons. Neurotropic viral infections are important causes of worldwide morbidity and death, and understanding how neurons have adapted to deal with VSV infections, given the selective pressure imposed upon the CNS due to spatial restrictions and difficulty replacing in neuronal networks, will facilitate identification of novel antiviral therapies.
Footnotes
Acknowledgments
We gratefully thank Dr. Gail Wertz (University of Virginia) for the generous gift of anti-VSV P and anti-VSV N polyclonal antibodies and Dr. Douglas S. Lyles (Bowman Gray Medical School) for the generous gift of the anti-M MAb 23H12. Ms. Jessica Amenta and Ms. Christina Griese contributed to some early experiments in this project. We also would like to thank Dr. Ignatius Tan (New York University) for helpful discussions and guidance in sample preparation; Dr. Chiye Aoki (New York University) for training, troubleshooting, and use of her electron microscope; as well as our colleague James M. Miller for providing additional editing and input. This work was supported by a grant (NS039746) from the NIH to C.S.R.
Disclosure Statement
No competing financial interests exist.