
Abstract
Neuroblastoma is a solid tumor of the sympathetic nervous system accounting for up to 10% of pediatric cancers and 15% of cancer-related deaths. It is a useful system for investigation of stress signal-mediated apoptosis as a tumor suppression mechanism. In this study, we present evidence that p53 mediates DNA damaging drug-induced apoptosis in IMR32 cells through the caspase-9 pathway. In summary, we define a molecular pathway for mediating DNA damaging drug-induced apoptosis in human neuroblastoma IMR32 cells and suggest that inactivation of essential components of this apoptotic pathway may confer drug resistance on neuroblastoma cells.
Introduction
The tumor suppressor p53 plays a pivotal role in mediating DNA damage-induced apoptosis (Rich et al., 2000; Hemann et al., 2005; Choi et al., 2008). The essential components of the signaling pathway that mediate the apoptotic response of p53 to DNA damage remain to be defined. Thymocytes and oncogene-transformed embryonic fibroblasts from caspase-9 null mice are resistant to irradiation and DNA damaging drug-induced apoptosis (Hakem et al., 1998; Cui et al., 2005), a p53-dependent process (Lowe et al., 1993a, 1993b; Hemann et al., 2005).
Neuroblastoma is a solid tumor of the sympathetic nervous system accounting for up to 10% of pediatric cancers and 15% of cancer-related deaths, which provides a particularly useful system for investigation of stress signal-mediated apoptosis as a tumor suppression mechanism. Several genetic abnormalities have been reported in neuroblastoma with the most common being MYCN amplification. Of all genetic aberrations identified, amplification of the oncogene MYCN remains a powerful prognostic determinant, indicating a poor prognosis, and is associated with remarkably decreased survival rates (Maris et al., 2007; Canete et al., 2009). This is clearly observed in cases of neuroblastoma where localized tumors or INSS stage 4S neuroblastoma that would otherwise be classified as tumors with a favorable outcome shift to the unfavorable, high-risk group requiring intensive multimodal therapy when MYCN amplification is present (Maris et al., 2007). MYCN amplification occurs in about 22% of neuroblastoma cases and is associated with advanced stages of the disease (Brodeur, 2003; Schwab et al., 2003; Jacobs et al., 2009). Thus, we chose a chemosensitive neuroblastoma cell line, IMR32, which has MYCN amplification, for our studies.
Neuroblastoma cells maintain a functional p53 signaling pathway (Goldman et al., 1996; Eggert et al., 2000; Keshelava et al., 2001; Cui et al., 2002, 2007; Meletis et al., 2006), and caspase-9, two critical components of the intrinsic pathway, are expressed and active in all neuroblastoma specimens examined (Teitz et al., 2002). Delineation of signaling pathways that mediate chemotherapeutic drug-induced apoptosis in chemosensitive neuroblastoma cells is essential to an eventual understanding of the mechanism underlying the poor response of late stage tumors to treatment. In this study, we address the question of how p53 mediates the DNA damaging drug-induced apoptosis in neuroblastoma.
Materials and Methods
Cell culture and cell lines
The human neuroblastoma cell line IMR32 (ATCC #CCL-127) and various cell lines derived from IMR32 were cultured in Dulbecco's modified Eagle's medium (Invitrogen) supplemented with 10% fetal bovine serum (Sigma). All cells were cultured at 37°C in a 5% CO2 humidified incubator.
Retroviral and plasmid constructs
The following retroviral and plasmid constructs were used to generate IMR32-derived cells for this study: pBabe/GFP retroviral vector was cloned by releasing the puromycin coding sequence with HinDIII and ClaI from pBabe-puro and replacing it with the enhanced green fluorescent protein cDNA, which was amplified by polymerase chain reaction from pEGFP-N1 (Clontech, Inc.). pSuper-puro/GFPsi contains a GFP-siRNA sequence under the control of the human H1-promoter. pBabe-puro/p53DN encodes a tumor-derived p53 mutant with arginine to histidine mutation at the residue 175, which functions as a dominant-negative mutant of p53 (Lindstrom et al., 2001). pSuper-puro/p53si contains a p53-siRNA sequence under the control of the human H1-promoter. pBabe-puro/casp9DN codes for a dominant-negative mutant of human caspase-9, in which the residue 287 is altered from cysteine to alanine (Srinivasula et al., 1998). Retroviruses were produced using the 293GPG packaging cell line as described (Ory et al., 1996). Briefly, 293GPG cells (∼1 × 107) were plated in a 10-cm dish, incubated for 24 h, and then transfected with 5 μg of a retroviral plasmid, using the LipofectAmine Plus protocol (Life Technologies, Inc.). Forty-eight hours after transfection, the retrovirus-containing medium was filtered through a 0.45 μm filter (Millipore) and supplemented with 4.0 μg/mL Polybrene (Sigma). For retroviral infections, IMR32 cells (∼2 × 106) were seeded in a 10-cm dish and incubated overnight. The cultured medium was then replaced by the retrovirus-containing medium. After 48 h, the viral supernatant was removed, and fresh culture medium containing 0.75 μg/mL puromycin was added. The cells were cultured in the selection medium for 3 days, and puromycin-resistant cells were pooled. The percentage of retrovirally infected cells ranged between 60% and 90%, as estimated in parallel infections using retroviruses expressing GFP. Expression of relevant genes was confirmed by functional and/or immunoblot analysis.
Apoptosis induction and analysis
Exponentially growing cells at 70%–80% confluence were either untreated or treated with the indicated concentrations of doxorubicin (Ben Venus Laboratories). After treatment, adherent and floating cells were pooled, collected by centrifugation, and washed once with ice-cold phosphate-buffered saline (PBS). Cell death was determined by trypan blue dye (0.2% in PBS). Apoptotic DNA fragments, devoid of most high molecular genomic DNA, were isolated and analyzed as described (Herrmann et al., 1994). Apoptotic cell death was determined by staining of the collected cells with annexin-V and 7-aminoactinomycin D, using a Guava cytometer according to the manufacturer's protocol (Guava).
Immunoblot analysis
Exponentially growing cells at 70%–80% confluence were untreated or treated with the indicated concentration or time course of doxorubicin. Cells were harvested at different time points after drug treatment and then washed with cold PBS. Cell pellets were suspended in sodium dodecyl sulfate sample buffer and boiled for 10 min. After centrifugation for 10 min, the sample obtained was electrophoresed on a 12% sodium dodecyl sulfate–polyacrylamide gel and transferred to nitrocellulose membrane. The film was then incubated with antibodies and visualized by ECL (Amersham Life Science Company). The following primary antibodies were used: a rabbit polyclonal antibody p53 at 1:500 dilution (FL-393; Santa Cruz), a mouse monoclonal antibody p21 waf1/cip1 at 1:250 dilution (clone SX118; Pharmingen Company), a mouse monoclonal antibody caspase-9 at 1:500 dilution (clone 96-2-22; upstate New York), and mouse monoclonal antibody α-tubulin at 1:2000 dilution (clone B-5-1-2; Sigma). Horseradish peroxidase labeled goat anti-rabbit or anti-mouse Group (ICN) antibodies were used as secondary antibodies.
Results
DNA damaging drugs induce rapid apoptosis in IMR32 cells
IMR32 neuroblastoma cells were treated with doxorubicin, a chemotherapy drug commonly used in the treatment of patients with neuroblastoma (Pizzo and Poplack, 1997). Cytotoxicity of doxorubicin was mainly due to its ability to induce DNA damage by inhibiting topoisomerase II activity (Wozniak and Ross, 1983; Tewey et al., 1984; Yang et al., 1985). As shown in Figure 1a, IMR32 cells were highly sensitive to doxorubicin, and more than half of the cell population lost viability within 24 h of treatment with either 0.05 or 0.1 μg/mL doxorubicin. This drug-induced cell death was apoptotic as indicated by the characteristic fragmentation of genomic DNA (Fig. 1b). Apoptotic cell death was further confirmed by an annexin-V binding assay in which annexin-V binds to externalized phosphatidylserine on the surface of apoptotic cells (Fig. 1c). These results suggest that IMR32 cells retain a functional signaling pathway for DNA damaging drug-induced apoptosis.
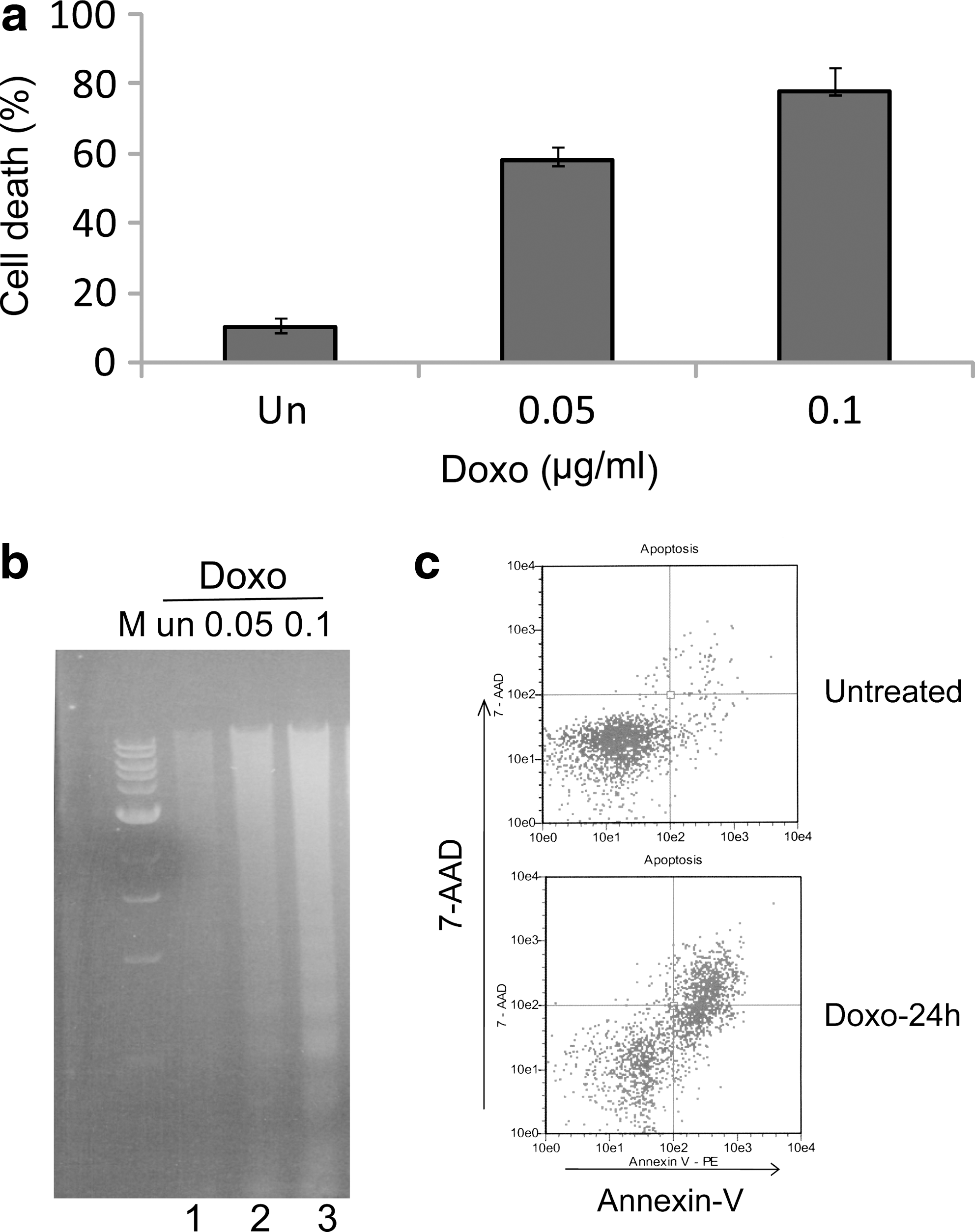
Doxorubicin induces apoptosis in IMR32 cells.
p53 signaling pathway is intact in IMR32 cells
We examined the functional status of p53 in IMR32 cells. Wild-type p53 protein has a short half life and is normally maintained at low, often undetectable levels in cells (Brown and Pagano, 1997). In response to DNA damage or other forms of genotoxic stress, p53 is stabilized, leading to a rapid increase in the p53 protein level and its activity. Activated p53, in turn, induces expression of its target genes, such as p21 waf1/cip1 (Barak et al., 1993; El-Deiry et al., 1993). Consistent with wild-type status, as shown in Figure 2, doxorubicin triggered a time-dependent increase of p53 protein, followed by the induction of p21. These results indicate that the p53 signaling pathway is functional in IMR32 cells.
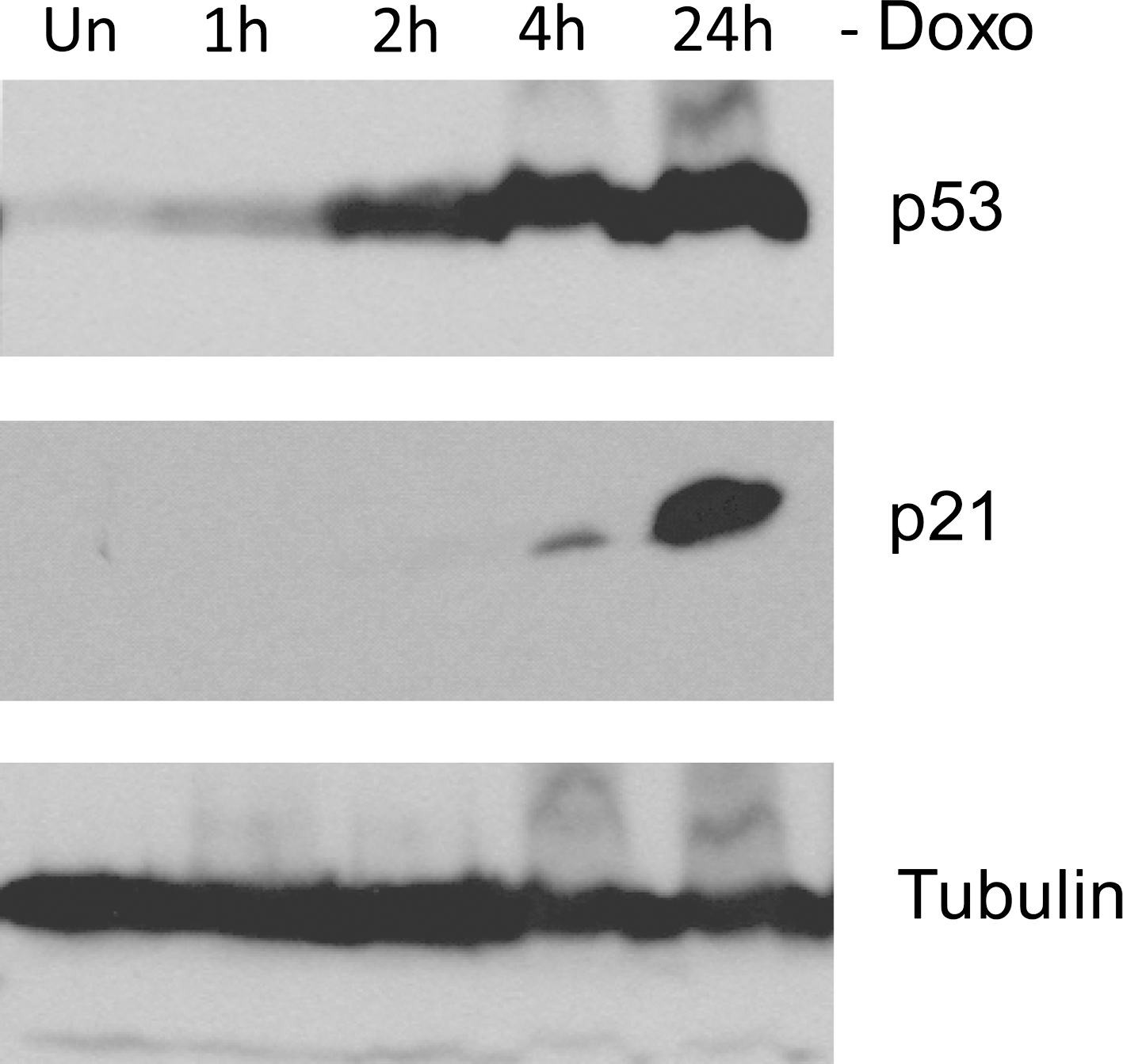
Activation of p53 signaling pathway in IMR32 cells. Immunoblot analysis of p53 and p21 expression in IMR32 cells at the indicated time points after doxorubicin (Doxo 0.5 μg/mL) treatment. α-tubulin levels are shown as loading controls.
p53 is required for DNA damaging drug-induced apoptosis in IMR32 cells
Next, we examined the contribution of p53 to DNA damaging drug-induced apoptosis in IMR32 cells by abrogating endogenous p53 activity. Retroviral-mediated gene transfer was used to introduce into IMR32 cells either p53si or the gene coding for a p53 dominant-negative mutant p53 (R175H) (p53DN). We detected the functional status of endogenous p53 in these cell lines by immunoblot analysis. p53 protein was barely detectable in p53si-expressing cells after the drug treatment, and no induction of p21 was observed (Fig. 3, right panel). The function of endogenous p53 expression was also significantly compromised in cells expressing p53DN, judged by the very low level of p21 induction in response to the drug (Fig. 3, left panel). In contrast, IMR32 cells expressing GFP showed normal p53 responses and the induction of p21 after drug treatment (Fig. 3, left and right panel) (GFPsi-expressing IMR32 cells showed similar results, data not shown). As expected, IMR32 cells expressing GFP remain highly sensitive to doxorubicin (Fig. 4a), and these cells were dying by apoptosis as indicated by the fragmentation of genomic DNA (Fig. 4b). However, IMR32 cells expressing either p53DN or p53si displayed remarkable resistance to the drug treatment (Fig. 4a) and failed to undergo apoptosis (Fig. 4b). According to these findings, we believe that p53 function is required for DNA damaging drug-induced apoptosis in IMR32 cells.
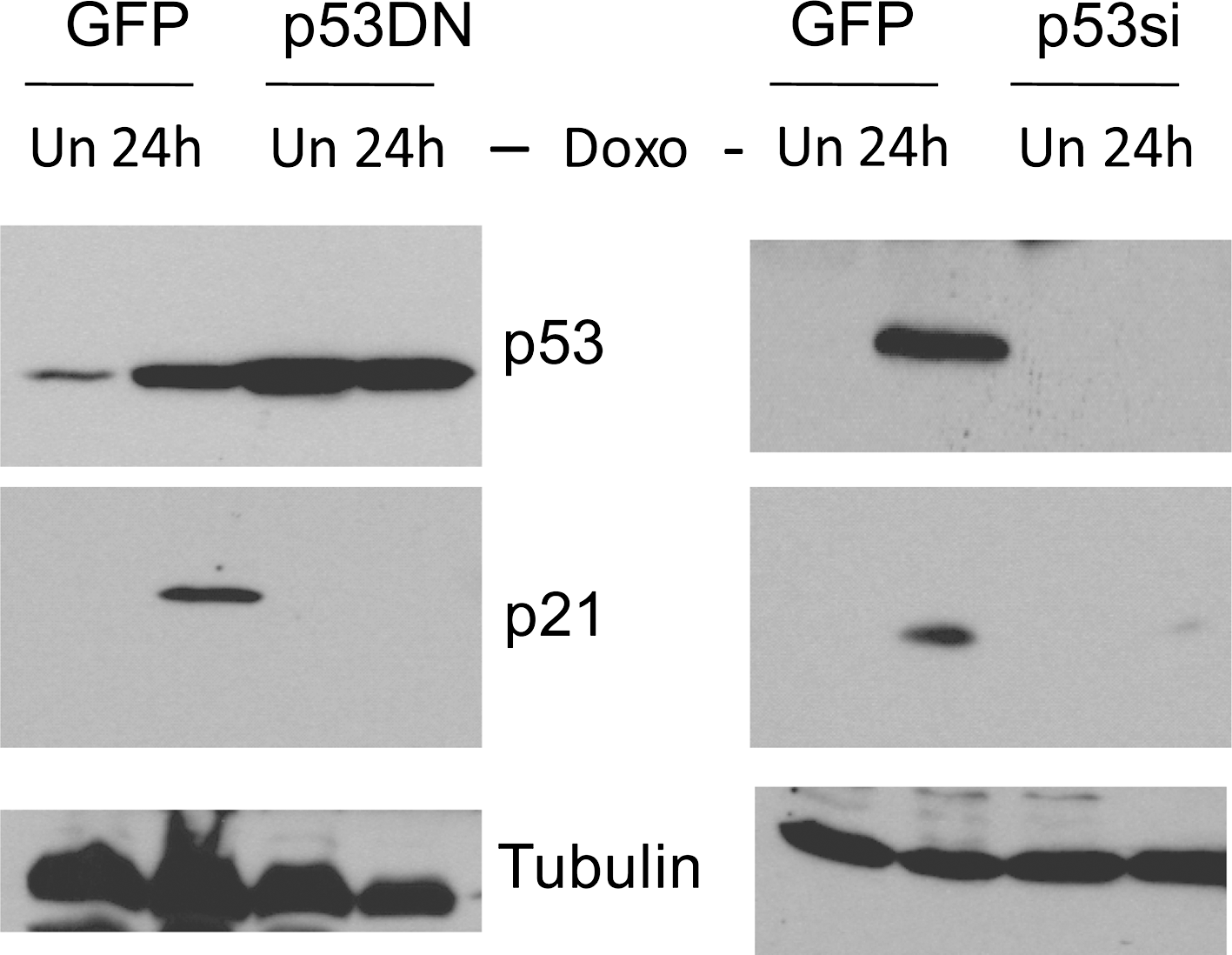
Inactivation of p53 by dominant-negative mutant p53 (p53DN) and p53si. Immunoblot analysis of p53 expression in GFP, p53DN, and p53si-expressing cells at the indicated time points after doxorubicin (Doxo 0.5 μg/mL) treatment. α-tubulin levels are shown as loading controls.
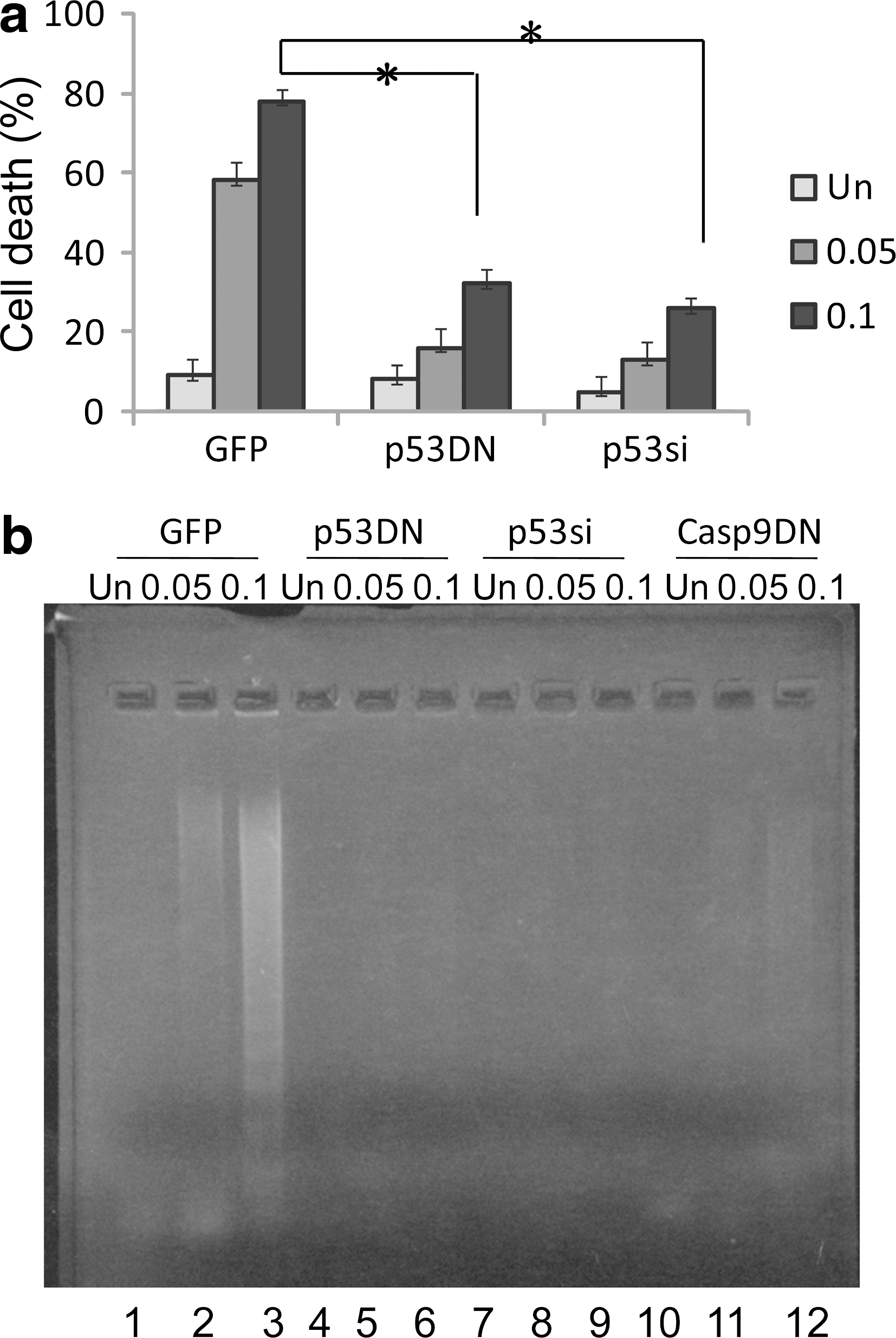
p53 is required in DNA damaging drug-induced apoptosis in IMR32 cells.
Caspase-9 is essential for mediating DNA damaging drug-induced apoptosis in IMR32 cells
Caspase-9 has been shown to play a major role in p53-dependent apoptosis (Hakem et al., 1998; Kuida et al., 1998). Activation of caspase-9 requires mitochondrial release of cytochrome c to the cytoplasm during apoptosis. We studied whether the mitochondrion/caspase-9 pathway was targeted by p53 in DNA damaging drug-induced apoptosis in IMR32 cells. Caspase-9 was indeed activated as indicated by the appearance of its processed forms, p35/p37 (Fig. 5a, left panel). However, activation of caspase-9 was totally blocked in IMR32 cells expressing caspase-9DN (Fig. 5a, right panel), which failed to induce cell death (Fig. 5b) and apoptosis (Fig. 4b, lanes 10–12). Expression of caspase-9DN was confirmed by immunoblot analysis (data not shown). The results suggested that the wild-type p53 targets the caspase-9 pathway for mediating DNA damaging drug-induced apoptosis in IMR32 cells.
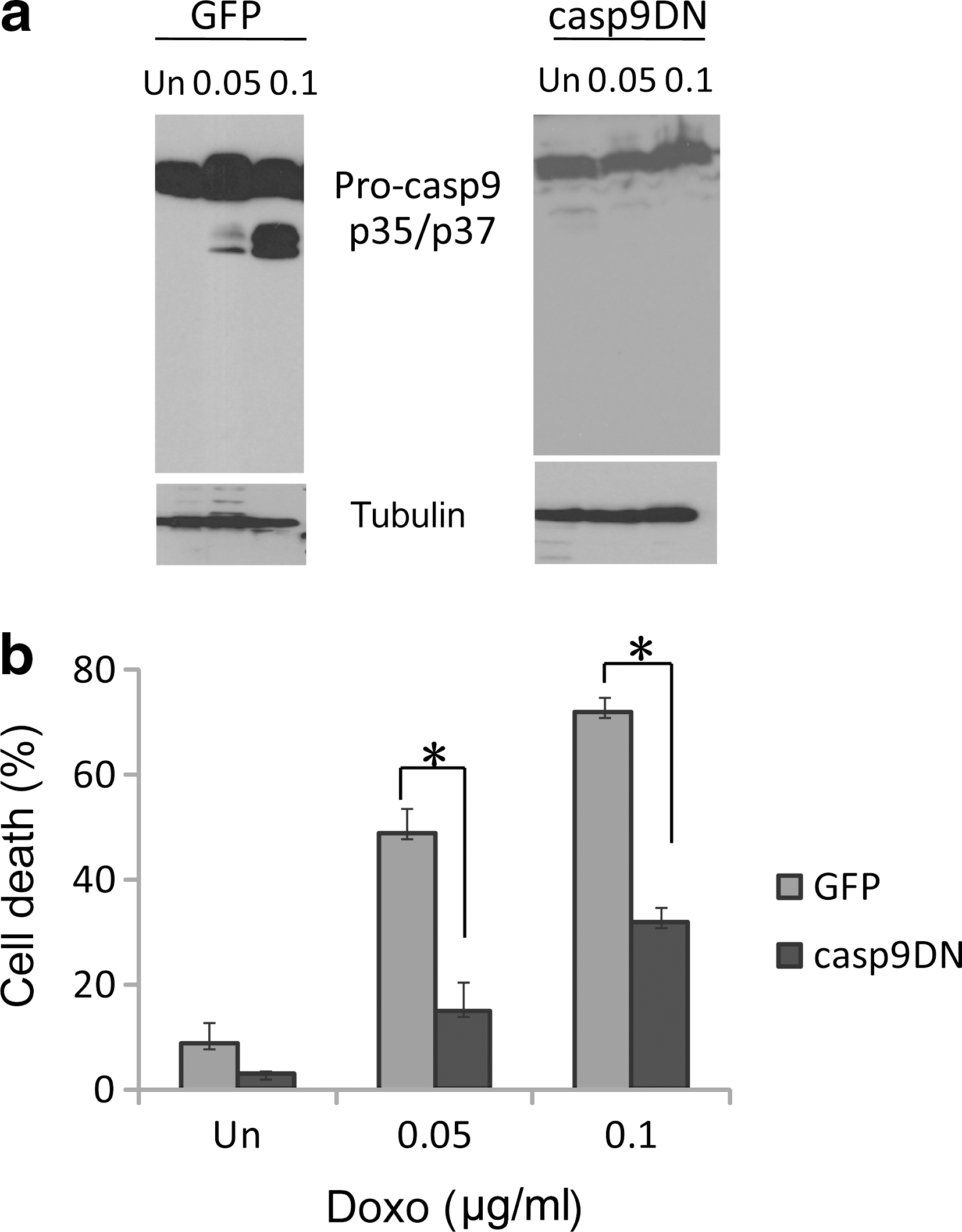
Caspase-9 is required in DNA damaging drug-induced apoptosis in IMR32 cells.
Discussion
Our results show that p53 and caspase-9 are required in DNA damaging drug-induced apoptosis in the chemosensitive IMR32 neuroblastoma cells which have MYCN amplification. We used the MYCN amplification cell line as a model to identify the mechanism responsible for neuroblastoma apoptosis. Currently, the criteria to determine risk stratification incorporate the age at diagnosis as well as the INSS stage, along with absence or presence of MYCN amplification, subsequently, placing patients in either the high-risk, intermediate-risk, or low-risk group (Schwab et al., 2003; Maris et al., 2007). p53 protein levels increase, leading to the induction of its transcription target p21. Inactivation of p53 by either p53si or p53DN completely protects IMR32 cells from drug-triggered apoptosis and blocks the caspase-9 activation (data not shown). In drug-treated cells, caspase-9 is activated. Over-expressing caspase-9DN can totally block cell death or apoptosis induced by the drug treatment. Unlike the majority of other human cancers in which p53 is commonly inactivated by mutations or other mechanisms (Hollstein et al., 1991; Vogelstein et al., 2000), neuroblastoma cells usually carry the wild-type p53 (Imamura et al., 1993; Komuro et al., 1993; Vogan et al., 1993; Castresana et al., 1994; Hosoi et al., 1994; Kusafuka et al., 1997). Our study determined an essential role of p53 in chemotherapeutic drug treatment. Similar results have been obtained from another two MYCN amplified neuroblastoma cell lines that were derived from the same patient before and after cytotoxic therapy. Tweddle et al. (2001) reported that cells derived before cytotoxic therapy carry wild-type p53 gene, but cells derived after 5 months at relapse have lost p53 function. The one with lost p53 function is far more resistant to irradiation and chemotherapeutic agents (Tweddle et al., 2001). However, it seems unlikely that the p53 signaling pathway is the only mediator of neuroblastoma cell apoptosis induced by DNA damaging drug.
Caspase-9 is a major mediator of p53-dependent apoptosis (Hakem et al., 1998; Kuida et al., 1998; Soengas et al., 1999). Our study shows that caspase-9 is also essential for the apoptotic function of p53 in IMR32 neuroblastoma cells. This implicates an important role for caspase-9 activity in conferring a favorable therapeutic index to neuroblastoma tumors during treatment. Thus, in defining a molecular pathway for mediating DNA damaging drug-induced apoptosis in a chemosensitive neuroblastoma cell line, our findings may aid in a molecular understanding of possible defects in signal transduction and regulation of this pathway in chemoresistant neuroblastoma cells.
In summary, our results define a molecular pathway for mediating DNA damaging drug-induced apoptosis in human neuroblastoma IMR32 cells and suggest that inactivation of p53 and caspase-9 may confer drug resistance on neuroblastoma cells.
Footnotes
Acknowledgments
We are grateful for Dr. Zhong-Huai Xiang and Dr. Han-Fei Ding for their invaluable support. We thank R. Mulligan for providing 293GPG cells, and J. Overmeyer and W. Maltese for providing IMR32 cells. Our work was supported by the Fundamental Research Fund for the Central Universities (No. XDJK2010A001).
Disclosure Statement
The authors are not employed by any commercial companies that may influence the study performed here.