
Abstract
Congenital heart defects are complicated birth defects due to the interaction of genetic and environmental factors. Previous research indicated the importance of transcription factors in heart development, which suggested that mutations of transcription factor genes could be genetic determinants of congenital heart defects. Recently, the length variation of an intronic region in the NFATC1 gene was linked to ventricular septal defect (VSD). In this study, we detected the length variation of the region in a Han Chinese population of patients with nonsyndromic VSD, atrial septal defect, patent ductus arteriosus, and control individuals. We found a new allele of the length variation with four repeats of a 44-bp region. At the same time, all the alleles were found in both patient and control groups and there were no significant differences in genotype distribution between the patients and controls. The results suggested no association of the length variation of the intronic region in NFATC1 gene with VSD, atrial septal defect, and patent ductus arteriosus.
Introduction
Embryonic heart development is a very complicated, evolutionarily conserved process, which implies the important role of the heart in animals (Srivastava and Olson, 2000). The regulation of this process involves many signaling molecules, especially transcription factors (Nemer and Nemer, 2001). Among these transcription factors, the nuclear factor of activated T cells (NFATC1) protein was first found in lymphocytes and is regulated by calcineurin, a Ca2+-dependent phosphatase (Serfling et al., 2000). Disruption of the NFATC1 gene in mice led to abnormal development of cardiac valves and septum as well as embryonic death (de la Pompa et al., 1998; Ranger et al., 1998). Further, several studies suggested that the NFATC1 protein was regulated by a network of vascular endothelial growth factor A, receptor activator of nuclear factor κB ligand, and calcineurin in cardiac valve morphogenesis (Chang et al., 2004; Combs and Yutzey, 2009).
Considering the important role of NFATC1 protein in cardiac valve development, mutations in the NFATC1 gene might be associated with CHDs. In 2006, Yehya et al. found that a differential duplication of an intronic region in the NFATC1 gene was associated with ventricular septal defect (VSD) (Yehya et al., 2006). Afterward, another study in Han Chinese people confirmed the association (Han et al., 2010). However, the relatively small numbers of patients and control individuals in these studies called for studies with more people to test their results. Therefore, we performed this study to test whether fragmental length variation of the intronic region in the NFATC1 gene was linked to nonsyndromic CHDs.
Materials and Methods
Subjects
All patients were recruited from the Division of Pediatric Cardiology in West China Second University Hospital, Sichuan University. They accepted interventional surgery for treating nonsyndromic CHDs, which were evaluated by pediatric cardiologists and diagnosed by echocardiography. Information about the patients is listed in Table 1. The parents of the patients were given a clear explanation of the potential risk of the study and signed the consent. The study was approved by the Ethics Committee of Sichuan University.
p-value from chi-square test comparing each patient group with the control group.
In the patient group, there are three individuals with both VSD and ASD and one person with both PDA and ASD. Thus, the total number of disease cases examined is four more than the number of patients in the study. Thus, the sum of patients classified by diseases is 133, 4 more than 129 (the amount of patients).
ASD, atrial septal defect; VSD, ventricular septal defect; PDA, patent ductus arteriosus.
Genotyping
DNA was extracted from white blood cells in whole blood by a human blood DNA isolation kit (Biomed, Beijing, China). Primers for PCR were the same as in a previous study (Yehya et al., 2006). Each PCR included 20 ng genomic DNA as template, 2.5 μL of 10X PCR buffer, 1.0 μL dNTP (2.5 mmol/L), 0.5 μL forward primer (10 pmol/μL), 0.5 μL reverse primer (10 pmol/μL), 0.125 μL rTaq polymerase (Transgen, Beijing, China), and distilled water to a total volume of 25 μL. Thermocycling parameters included an initial denaturation of 5 min at 95°C; 35 cycles of 95°C for 30 s, 61.5°C for 30 s, and 72°C for 1 min; and a final extension at 72°C for 10 min. PCR products were detected by electrophoresis on 3% agarose gel. Three bands corresponding to different lengths were cut, purified, and sequenced using an ABI 3730 automated sequencer (Applied Biosystems, Foster City, CA). We replicated PCR and detection of 30% of the samples, ensuring that replicates were performed by a different researcher. Ambiguous results were confirmed by another PCR and electrophoresis analysis.
Statistics
Power of this study was calculated by Quanto v1.2.4 (Gauderman, 2002; Gauderman and Morrison, 2006). The allele distribution of the fragment variation was compared by chi-square test in different groups. A two-tailed p was used and the cutoff value was 0.05. The statistic analyses and histogram drawing were performed by SPSS software package (SPSS version 16.0, Chicago, IL).
Results
One hundred twenty-nine children with CHDs, including VSD, atrial septal defect (ASD), and patent ductus arteriosus (PDA), were recruited in this study. Among them, 60 individuals were affected by isolated VSD, 46 by isolated PDA, 18 by isolated ASD, 3 by both VSD and ASD, 1 by both pulmonary stenosis and ASD, and 1 by both persistent truncus ateriosus and ASD. The power calculation suggested that the sample size of this study can detect an OR of >1.95, and for subgroups, the sample size of VSD, PDA, and ASD can detect ORs of >2.55, 3.00, and 4.95, respectively.
We analyzed duplications in the intronic region between exons 7 and 8 of the NFATC1 gene in all the patients and control children. In our population, we found three different alleles, resulting in five distinct patterns of bands on agarose gel. The three alleles were the wild-type allele (O), one with an insertion of a 44-bp segment (I1), and a novel one with an insertion of an 88-bp segment (I2). As far as we know, the I2 allele has not been reported previously. By sequencing, we found that the 88-bp insertion was a duplication of the 44-bp segment (Supplementary Fig. S1; Supplementary Data are available online at
To check whether an association exists between the fragment length variations and CHDs, we amplified the target fragments of all the patients with VSD and ASD and compared the distributions of different genotypes in patient groups to the results of the 179 control children (Fig. 1). As shown in Table 1, there were no differences of genotype distributions between patient and control groups. There was only one child with the I1/I2 genotype in the whole population, suggesting that this genotype is rare, and it was excluded from further analyses.
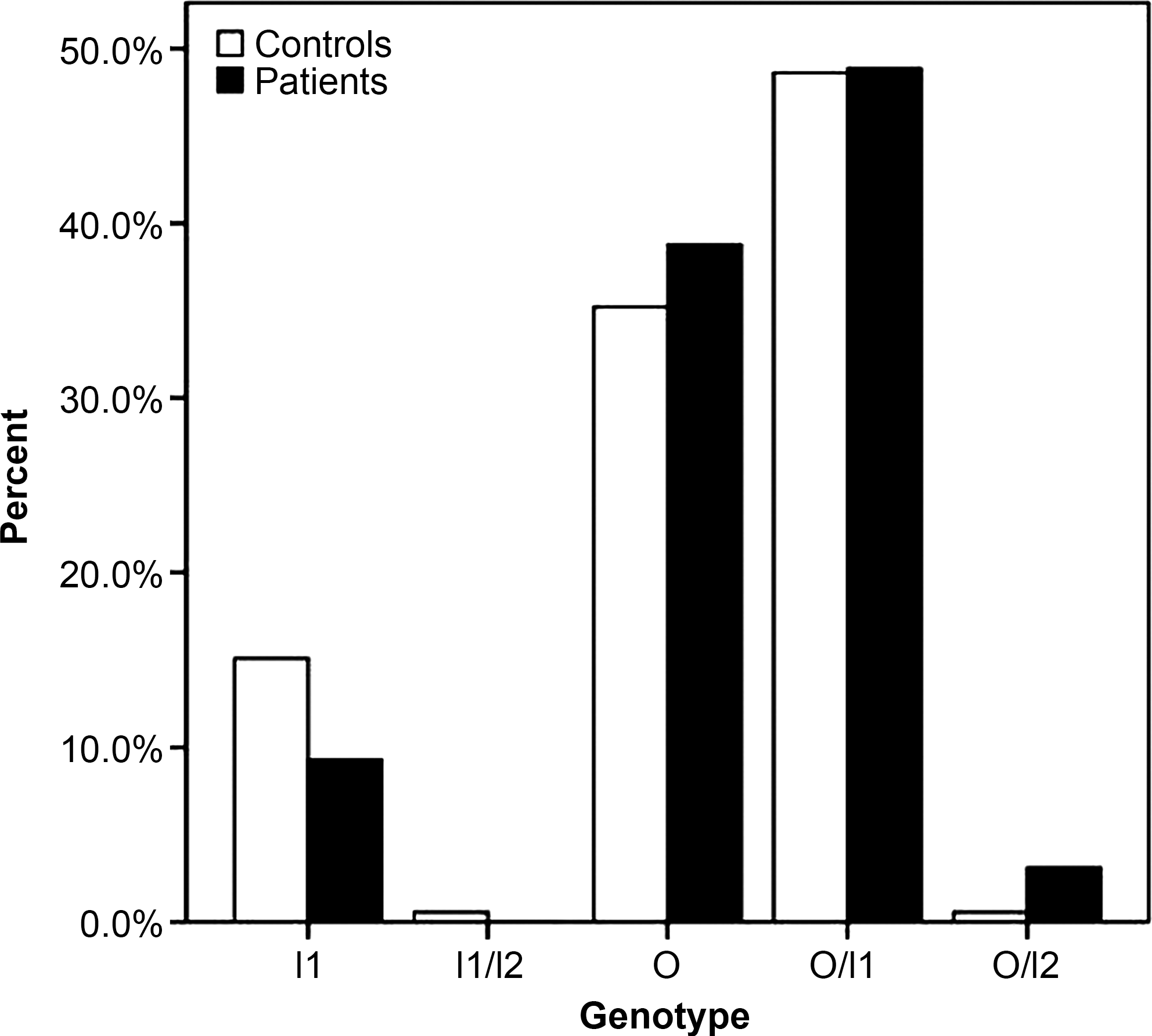
Comparison of the genotype distributions of the intronic region in patients and controls.
Discussion
CHDs are a high prevalence defects with an occurrence of about 7 in 1000 live births (Hoffman and Kaplan, 2002). Sporadic nonsyndromic CHDs are thought to be influenced by genetic and environmental interactions (Wessels and Willems, 2010). Cooperation among mutations insusceptibility genes and environmental factors was hypothesized to explain the sporadic nature of nonsyndromic CHDs (Burn et al., 1998). Although this hypothesis was widely accepted, only a few studies on the interactions were performed. On the other hand, genetic studies have implicated >40 different genes involved in nonsyndromic CHDs [reviewed by Wessels and Willems (2010)]. However, the genetic mechanisms of nonsyndromic CHDs in most patients remain unclear.
In mouse studies, the transcription factor NF-ATc was associated with morphogenesis of cardiac valves and septum and suggested as a candidate molecule involved in CHDs (de la Pompa et al., 1998; Ranger et al., 1998). In 2006, a report showed that the duplication of an intronic region in the NFATC1 gene was associated with VSD (Yehya et al., 2006). However, the population size was limited in their study. In this study, we scanned this region in 129 children with nonsyndromic CHDs and 179 children with no heart defects. There were no significant differences in genotype distribution between affected children and control children. Yehya et al. studied only 81 unaffected subjects and they did not find I1 allele homozygotes. However, in this study, we found that I1 allele homozygotes existed in the control group with a relatively high frequency (27/179, 15.84%). Considering that there was no clear description of racial data in the study by Yehya et al., the discrepancy can be explained by the relatively few control individuals in their study and the different ethnic populations in the two studies. Recently, a study in a Han Chinese population confirmed that I1 allele homozygotes appeared only in patients with VSD, ASD, and bicuspid aortic valve (Han et al., 2010). As the control individuals in their study were patients with heart disease not CHDs, they could not describe the frequency of the I1 allele in the normal population. This study, on the basis of a larger patient group and control group, suggested no association between the fragmental polymorphism in intron 7 of the NFATC1 gene and VSD, PDA, and ASD. In addition, we found a new allele of this polymorphism, which includes four repeats of the 44-bp sequence. The result underlined the complicated polymorphism of this fragment. Future studies with an even larger population are needed to elucidate whether this polymorphism is functional.
Footnotes
Acknowledgments
The authors are grateful to the people who participated in this research. This work was supported by National Basic Research Program of China (No. 2007CB511905) and the Fundamental Research Funds for the Central Universities (No. 2010SCU3003).
Disclosure Statement
The authors declare that there are no conflicts of interest.
References
Supplementary Material
Please find the following supplemental material available below.
For Open Access articles published under a Creative Commons License, all supplemental material carries the same license as the article it is associated with.
For non-Open Access articles published, all supplemental material carries a non-exclusive license, and permission requests for re-use of supplemental material or any part of supplemental material shall be sent directly to the copyright owner as specified in the copyright notice associated with the article.