
Abstract
The processing bodies (PBs) are a form of cytoplasmic aggregates that house the cellular RNA decay machinery as well as many RNA-binding proteins and mRNAs. The PBs are constitutively present in eukaryotic cells and are involved in maintaining cellular homeostasis by regulating RNA metabolism, cell signaling, and survival. Virus infections result in modification of the PBs and their constituents. Many viruses induce compositionally altered PBs, while many others use specific components of the PBs for their replication. PB constituents are also known to restrict virus replication by a variety of mechanisms. Further, continuing studies in this rapidly emerging field of PB-virus interactions will undoubtedly provide important clues to the understanding of the role of PBs in cellular homeostasis as well as their role in virus infections and innate immune signaling.
Introduction
The PBs are constitutively present in the cytoplasm of cells under physiological conditions (Kulkarni et al., 2010) and their number and size increase upon translational arrest (Teixeira et al., 2005). The PBs are involved in sequestration, storage, and/or degradation of mRNAs (Brengues et al., 2005; Kulkarni et al., 2010). Constituents of PBs include proteins that function in general mRNA degradation pathways, such as XrnI, Dcp1-2, Lsm1-7, Lsm16 (Edc3), Rap55 (Lsm14A), Ge1 (Hedls, Edc4), PatL1, DDX6; proteins involved in nonsense-mediated mRNA decay (NMD) pathway; translational repressors, such as eIF4E-T, CPEB1, Lsm14A, PatL1, and DDX6; and components of the RNAi pathway, including Ago1-4, GW182, Dicer, miRNAs (Ernoult-Lange et al., 2012). The PBs and SGs share many mRNAs and proteins (described here as PB-SG markers) such as Ago1-4, APOBEC, BRF1, DDX3/6, eIF4E, FAST Lin28, Mex3b, PCBP1-2, Rap55, TTP, Upf1, Xrn1; however, proteins, such as Ccr4, Dcp1-2, Edc1-3, eIF4E-T, GW182, Ge1, Lsm1-7, Mex3a, PatL1, Pan2, Pop2 (described here as PB markers) are only found in the PBs (Buchan and Parker, 2009) (Fig. 1). It has been suggested that the PBs are the nucleation sites for SG formation (Kedersha et al., 2005).
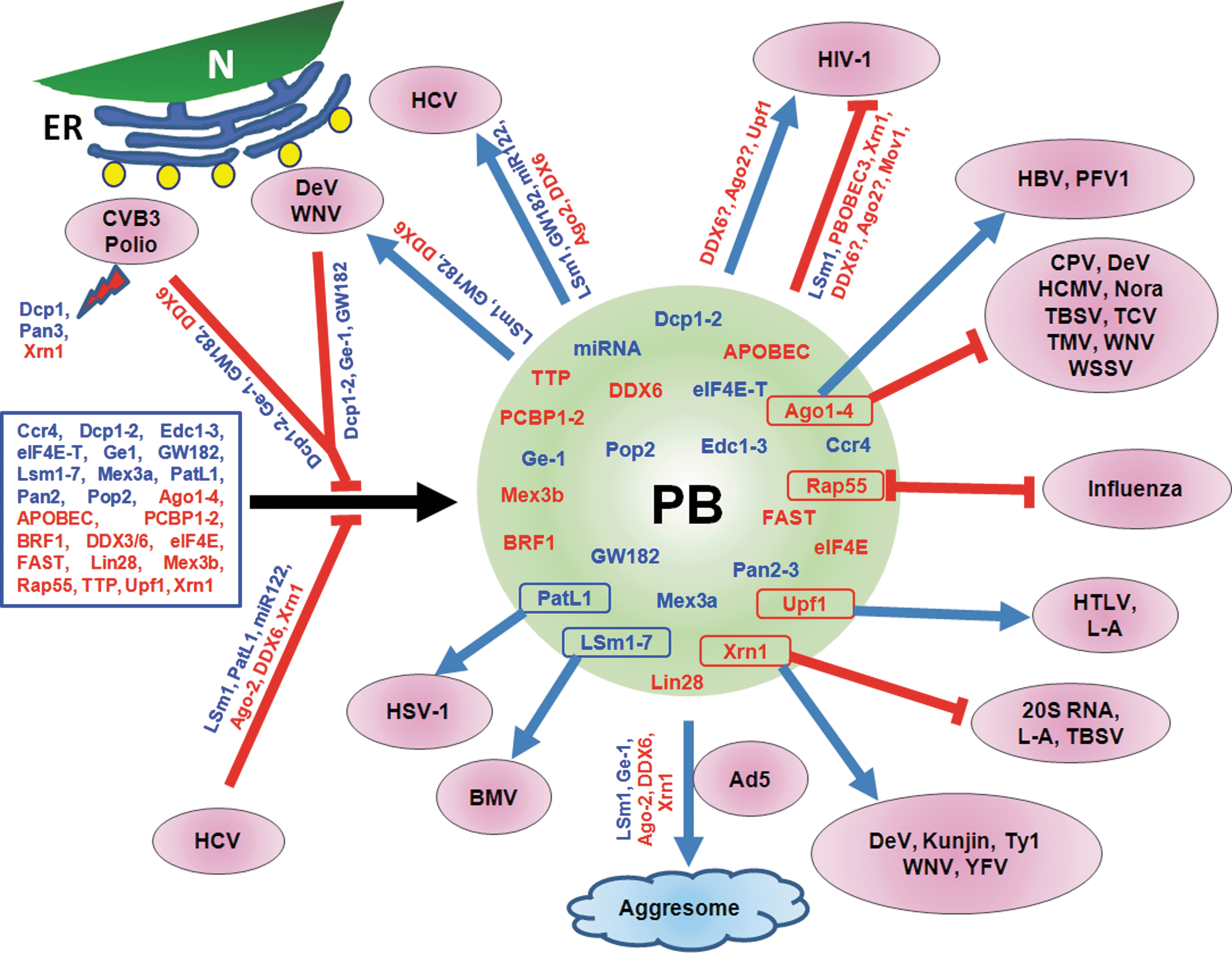
Interaction of viruses with PBs and its constituents. Cellular proteins (in rectangular box on the left side) aggregate to form PB (shown as a circle) with PB markers in blue and PB-SG markers in red. Shown are viruses that use various PB constituents to support their replication, while other viruses are inhibited by various components. HCV, WNV, DeV, Polio, and CVB3 virus infection cause redistribution of PB markers to viral replication sites near lipid droplet (yellow circles) around the ER. Some viruses inhibit aggregation of proteins (identified along the lines) into PB. Many individual PB and PB-SG components (identified with outlines within the circle) have been found to support or antagonize virus replication. SG, stress granule; PBs, processing bodies; HIV-1, human immunodeficiency virus 1; HBV, hepatitis B virus; PFV1, prototype foamy virus 1; TMV, tobacco mosaic virus; TBSV, tomato bushy stunt virus; CPV, cricket paralysis virus; WSSV, white spot syndrome virus; TCV, turnip crinkle virus; HCMV, human cytomegalovirus; DeV, dengue virus; WNV, West Nile virus; HTLV, human T-lymphotropic virus; L-A, yeast L-A double-stranded RNA virus; 20S RNA, 20S RNA narnavirus; YFV, yellow fever virus; Ty1, Ty1 retrotransposon; Ad5, adenovirus 5; BMV, brome mosaic virus; HSV-1, herpes simplex virus 1; HCV, hepatitis C virus; CVB3, Coxsackie virus B3; ER, endoplasmic reticulum; N, nucleus.
Alteration of PBs by Virus Replication
Although a significant body of information is available on how viruses influence the SGs and their constituents, less is known about the viral manipulation of the PBs and their constituents. Interestingly, the majority of the viruses currently known to influence PBs are the RNA viruses, most of which are positive-stranded.
Recent studies have revealed that HCV replication leads to dispersion or alteration of PBs in cells, while virus-receptor interactions or viral protein expression have no discernible effect (Ariumi et al., 2011; Perez-Vilaro et al., 2012; Pager et al., 2013). Interestingly, not all PBs were disrupted by HCV replication (Perez-Vilaro et al., 2012; Pager et al., 2013) and the number of PBs as examined by the use of PB markers Dcp1 or GW182 were similar to those in uninfected cells (Perez-Vilaro et al., 2012), indicating that HCV replication alters PB composition (Perez-Vilaro et al., 2012; Pager et al., 2013). Similarly, in cells infected with West Nile virus (WNV) and Dengue virus, PBs are disrupted and their number declines due primarily to the recruitment of many of the PB constituents (LSm1, GW182, DDX3/6, Xrn1) to the sites of viral replication (Emara and Brinton, 2007; Chahar et al., 2012). Disruption of PBs is also seen in cells infected with poliovirus and Coxsackievirus B3 as a result of degradation of Pan3, Xrn1, and Dcp1 (Dougherty et al., 2011), although other PB markers (DDX6, GW182, Edc3) were not degraded. It appears that degradation of some PB markers in these picornavirus-infected cells may be sufficient to disrupt PBs. However, use of multiple PB markers would be necessary for a clear understanding of the role of PBs in these virus-infected cells.
Among the negative-strand RNA viruses, the influenza virus NS1 protein suppresses formation of PBs (Mok et al., 2012) through interaction with Rap55, a PB constituent. This interaction is thought to be necessary for efficient viral replication through blocking the sequestration of the viral NP and the NP-associated RNAs in PBs. The PBs also appears to play important role(s) in supporting Hantanvirus transcription by sequestering nucleocapsid protein-bound cellular mRNAs containing 5′-caps (Mir et al., 2008). In vesicular stomatitis virus (VSV) infected cells, viral P protein interacts and colocalizes with PB-SG markers, PCBP1/2 (Dinh et al., 2011). Although PB formation appears to be unaffected by VSV infection (Dinh et al., 2013), further work with additional PB-specific markers would be necessary.
Less is evident about how DNA viruses affect PBs in infected cells. The number of PBs is reduced in adenovirus-infected cells as the viral E4 11K protein, through its specific interaction with DDX6, mediates redistribution of several PB components to aggresomes (Greer et al., 2011) (Fig. 1), the sites for inactivation or degradation of cellular misfolded/unwanted proteins (Kopito, 2000).
Thus, it appears that individual viruses manipulate and modify the PBs and their constituents by disparate mechanisms; however, the precise role(s) of the PBs in viral life cycle remains unclear. This is primarily due to the fact that (1) majority of these studies use PB-SG markers but draw specific conclusions only for PBs; (2) in many cases, the number of PB markers used is limited, especially when a PB protein is silenced, PB formation by that specific protein was examined, while PB formation by other authentic PB markers was not analyzed; and (3) in several studies, the role of PB markers hijacked by viruses was not investigated simultaneously with the disruption of PB. These experimental shortcomings have restricted unequivocal interpretation of the role of the PBs and/or their constituents in virus infections.
PB Constituents That Support Virus Replication
Among the PB components translocating to HCV replication sites around lipid droplets, DDX6, Ago2, Lsm1, PatL1, Ge1, and miR122 were found to be required for HCV replication as their knockdown negatively affected viral RNA replication and virus growth (Ariumi et al., 2011; Berezhna et al., 2011; Pager et al., 2013; Thibault et al., 2013). In contrast, knockdown of Rap55, another PB-associated marker, resulted in reduced PBs in the cells, but had no adverse effect on HCV replication or virus production (Perez-Vilaro et al., 2012). HCV replication supporting structures formed around lipid droplets that contain hijacked PB markers appear to be distinct from the canonical PBs, suggesting that HCV replication induces the formation of compositionally altered PBs (Berezhna et al., 2011; Perez-Vilaro et al., 2012). It appears that HCV selectively takes advantage of several factors belonging to PB-RNAi pathway (Ago2, GW182, miR122) and PB-mRNA decay pathway (LSm1, GW182, DDX6, Ge1, Xrn1) for replication, while other PB components, such as Rap55, Dcp1-2 do not seem to influence HCV replication. Mechanistically, how the PB constituents that translocate to lipid droplets are involved for HCV replication is not known at this time but a protective role of miR122 in antagonizing the exonuclease activity of Xrn1 in HCV RNA degradation has been recently demonstrated (Li et al., 2013).
As shown in Figure 1, PB constituents support replication of many viruses in mammalian cells. PB constituents are also required for virus infections in yeast. Using Saccharomyces cerevisiae, Dhh1 (DDX6 homolog), Kem1 (Xrn1 homolog), LSm1, and Pat1 (PatL1 homolog) were shown to support assembly of retrotransposition-competent virus-like particles of Ty1 (Checkley et al., 2010). Brome mosaic virus (BMV) infection increases the number and size of PB foci (Beckham et al., 2007) and it has been shown that BMV replication and protein expression in yeast requires LSm1-7, Dhh1, and Pat1. LSm1 interacts and colocalizes with viral RdRp and RNA. LSm1-7 binds to a tRNA-like structure at the 3′-untranslated region and two internal A-rich single-stranded regions in the BMV genome to regulate viral replication and translation (Galao et al., 2010).
Utilization of PB components by DNA viruses is less well characterized. HSV-1 Us11 protein was shown to interact with PatL1, a cellular kinesin light-chain-related protein (Benboudjema et al., 2003). This interaction is presumed to be important for the intracellular movement of viral components. Hepatitis B virus (HBV), core (HBc), and surface (HBs) proteins but not HBx protein interact and colocalize with Ago2 in the ER and other subcellular compartments, while Ago2 appears to play a role in HBV life cycle (Hayes et al., 2012). The Kaposi's sarcoma-associated herpesvirus SOX protein induces internal cleavage of host mRNA, which are then degraded by the 5′-3′ cellular exonuclease Xrn1 (Covarrubias et al., 2011).
These studies shed light into our understanding of how viruses exploit PB proteins to enhance their replication. Many components of the cellular RNAi pathway as well as miRNAs are localized into PBs and it seems that in addition to its putative roles in suppression of virus infection (Arbuthnot, 2010), multiple PB-RNAi proteins are utilized by both RNA and DNA viruses for their replication, assembly and budding.
PB Constituents That Suppress Virus Replication
PB constituents are also known to suppress replication of many viruses (Fig. 1). Prominent among these are certain members of APOBEC3 (apolipoprotein B mRNA editing enzyme catalytic polypeptide like 3) family, which are incorporated into PBs and encode cytidine deaminase activity to mutate cytidine residues into uridines in nascent minus strand of retroviral reverse transcripts leading to G to A hyper-mutation in the plus-strand of viral DNA and compromise virus replication (Harris et al., 2003; Bishop et al., 2004). Among the seven members in APOBEC3 family, four have been shown to antagonize HIV-1 (Martin et al., 2011; Imahashi et al., 2012). APOBEC3F and 3G localize to PBs as well as SGs (Wichroski et al., 2006; Gallois-Montbrun et al., 2007) where they interact with different proteins, including the RISC protein (Ago2), PB proteins, (Dcp1 and Dcp2) and PB-SG protein (DDX6) (Kozak et al., 2006). Although discrepancies exist with regard to interactions among various PB constituents and HIV-1 RNA, mRNA, and Gag proteins (Nathans et al., 2009; Yu et al., 2011; Bouttier et al., 2012; Phalora et al., 2012; Reed et al., 2012), the majority of the studies show that many PB components suppress HIV-1 replication by multiple mechanisms. Similar to APOBEC3, Mov10, an RNA helicase, is incorporated into HIV-1 particles. Mov10 associates with APOBEC3, localizes to PB as a partner of Ago2 in RNAi pathway (Meister et al., 2005) and inhibits HIV-1 replication (Burdick et al., 2010).
Viral Countermeasures for PB Constituents
The antagonistic properties of APOBEC3 proteins on HIV-1 replication are countered by the viral vif protein (Imahashi et al., 2012), which degrades APOBEC3 proteins. In human T-lymphotropic virus type 1 infection, viral tax protein inhibits NMD pathway via upregulation of Upf1 phosphorylation and accumulation of phospho-Upf1 in PBs, leading to stabilization of viral and cellular mRNAs to support virus infection (Mocquet et al., 2012). Another PB constituent that is targeted by a viral component is Xrn1, the 5′-3′ exonuclease, which degrades decapped mRNAs (Jones et al., 2012). Genomic RNAs of positive stranded and double stranded RNA viruses serve directly as mRNA for translation and their stability is regulated by Xrn1 (and its homologs) as demonstrated for HCV, Tombusvirus, and L-A virus (Esteban et al., 2008; Jaag and Nagy, 2009; Li et al., 2013). In contrast, Xrn1-mediated mRNA cleavage is not allowed to complete the degradation of viral genome RNA in cells infected with Flaviviruses (including WNV, Dengue, Kunjin, and Yellow fever virus) (Funk et al., 2010; Silva et al., 2010; Moon et al., 2012) due to the presence of secondary and tertiary structures at the 3′-UTR of the viral genomes, resulting in the production of short subgenomic flavivirus RNA (sfRNA), which is required for viral infections (Moon et al., 2012) and pathogenesis in mice (Silva et al., 2010). These studies indicate that sfRNA attenuates Xrn1 activity to allow virus replication to continue unabated. In contrast, Poliovirus selectively degrades Xrn1, a step presumed to be prerequisite for subsequent disruption of PB in infected cells (Dougherty et al., 2011).
Concluding Remarks
The consequences of viral interactions with cellular PBs and their constituents appear to be multifold. While some are beneficial, other interactions negatively affect virus replication, and therefore, viruses have evolved mechanisms to counter the PBs and their components. As the field of PB-virus interactions is growing rapidly, mechanistic understanding of how different viruses manipulate PBs and their constituents will likely emerge. The current knowledge of the role of PBs in virus infections is less complete primarily due to lack of studies using multiple PB-specific markers in infected cells. Thus, the real role(s) played by PBs in virus infections remain elusive. Additionally, how virus infections and subsequent cellular signaling events lead to increase in size and number of PBs in cells is poorly understood. Since PBs are intracellular granules constitutively present in normal cells and play key roles in RNA metabolism, studies examining how viruses modulate DYRK3-mTORC1 signaling in granule formation and dissolution may provide a better understanding of the form and function of this intriguing organelle in cell biology as well as in virus-host interactions. Recent identification of Rap55, a PB-associated protein, as a sensor of viral nucleic acids that activates antiviral responses in early stages of viral infections (Li et al., 2012) suggests that PBs and their constituents are not only important maintaining cellular homeostasis by regulating RNA metabolism, cell signaling, and survival but they may also play key roles in innate immune signaling.
Footnotes
Disclosure Statement
No competing financial interests exist.