
Abstract
Common genetic variants (single-nucleotide polymorphisms [SNPs]) in microRNA genes may alter their maturation or expression, resulting in varied functional consequences. Several studies have evaluated the association between the SNP rs11614913 and cancer risk in diverse populations and in a range of cancers, with contradictory outcomes. In this study, we examined 114 paired samples (tumor and normal tissues) from breast cancer patients to study the genotype distribution and somatic mutation of the SNP in MIR 196A2 (rs11614913 C-T). In addition, we evaluated their influence on the mature MIR 196A2 expression. We found that 14% (16/114) of tumors underwent somatic mutation of the SNP rs11614913. Moreover, the CT heterozygous and the CC homozygous states of SNP rs11614913 were more prone to mutation, while the TT homozygous state appeared to be resistant. We further detected a significant increase (p = 0.002) in mature MIR 196A2 expression in breast cancer. In particular, we found a significant association between the occurrence of SNP rs11614913 mutation and high expression (p = 0.0002). In addition, the mature MIR 196A2 expression level was significantly associated with the higher tumor grade (p = 0.004). Taken together, our results seem to demonstrate that somatic mutation of SNP rs11614913 in MIR 196A2 can have an influence on its expression. In addition, it indicated that an unknown mechanism is responsible for both the mutation of SNP rs11614913 and the dysregulation of mature MIR 196A2 expression.
Introduction
M
MIR 196A2 was highly expressed in breast (Iorio et al., 2005; Hui et al., 2009) and pancreatic (Bloomston et al., 2007) cancers compared with normal tissues, and the elevated expression was associated with significantly reduced survival for pancreatic cancer (Bloomston et al., 2007). Recent data have shown that SNP rs11614913 may influence the expression of mature MIR 196A2 (Hu et al., 2008; Hoffman et al., 2009). SNP rs11614913 was initially reported to be a prognostic biomarker for nonsmall cell lung cancer by Hu et al. (2008). Since then, the association between the rs11614913 polymorphism and susceptibility to diverse types of human cancer has been analyzed in several molecular epidemiological studies (Horikawa et al., 2008; Yang et al., 2008; Hoffman et al., 2009; Hu et al., 2009; Tian et al., 2009; Catucci et al., 2010; Christensen et al., 2010; Li et al., 2010; Qi et al., 2010; Srivastava et al., 2010; Akkiz et al., 2011; Hong et al., 2011). However, the results are generally controversial.
In the present work, we identified that a functional variant rs11614913 in MIR 196A2 was frequently mutated in breast cancers. Particularly, analysis of the mutations revealed that the CT heterozygous and the CC homozygous states were more prone to mutation, while the TT homozygous state appeared to be resistant. In addition, we found that mature MIR 196A2 expression level was elevated in breast cancers. Interestingly, the occurrence of rs11614913 mutation was significantly associated with the higher expression level of MIR 196A2 in the tumors. In addition, the mature MIR 196A2 expression level was significantly associated with the higher tumor grade.
Materials and Methods
Cell lines, patients, and tissue samples
Ten breast epithelial cell lines were used in this study, including eight breast cancer cell lines (BT-549, HCC1806, Hs578T, MCF7, MDA-MB-231, SW527, T-47D, and ZR-75–1) and two immortalized but nonneoplastic breast epithelial cell lines (184A1 and MCF10A). All the cell lines were purchased from the American Type Culture Collection (Manassas, VA). A total of 114 paired breast cancer samples were used. Fresh breast cancer tissues and adjacent normal tissues were collected after surgery, snap frozen in liquid nitrogen, and stored at −80°C until use. All tissue specimens were obtained from the surgical treatment of patients with breast cancer at the Cancer Hospital of Tianjin Medical University between May 2008 and March 2009. The use of the materials was approved by the Ethics Committee of the Cancer Hospital of Tianjin Medical University.
DNA extraction and sequencing
Genomic DNA from fresh cancer tissues was extracted using the DNeasy Blood & Tissue Kit (QIAGEN, Shanghai, China). DNA was then amplified by PCR using primers 5′-CCCCT TCCCTTCTCCTCCAGATA-3′ (forward) and 5′-CGAAA ACCGACTGATGTAACT-CCG-3′ (reverse) for the SNP. PCR products were then purified and sequenced (Invitrogen, Beijing, China). When a sequence alteration was detected, PCR and sequencing were repeated to confirm.
Detection of MIR 196A2 expression by real-time RT-PCR
To evaluate the expression of MIR 196A2 in cancer specimens carrying differential genotypes of SNP rs11614913, we examined 43 paired normal and breast cancer tissues, including 10 cases with the CC genotype, 10 with the CT genotype, 10 with the TT genotype, and 13 cases that had somatic mutations in MIR 196A2 (the RNAlater-treated tissues of the other three cases that had somatic mutations were used). Stem-loop real-time reverse transcription–polymerase chain reaction (RT-PCR) was used. Total RNA was extracted from 43 paired normal and breast cancer tissues using the TRIzol reagent (Invitrogen, Shanghai, China) according to the manufacturer's instructions. The reverse transcription was carried out with the TaqMan Reverse Transcription Kit (Invitrogen), and the expression of MIR 196A2 was determined using the TaqMan microRNA Assay (Invitrogen) in the Mastercycler ep realplex PCR thermal cycler (Eppendorf, Shanghai, China). The U6 RNA was used as an endogenous control (Invitrogen).
Statistical analyses
The association between the genotypes of the SNP rs11614913 and clinicopathological variables in normal and tumor tissues was determined using the χ2 test. The mean levels of MIR 196A2 expression in tumor tissues and paired adjacent normal tissues were analyzed using the t-test. The difference in relative expression of MIR 196A2 by SNP rs11614913 genotype was evaluated by ANOVA. The association between the levels of MIR 196A2 expression with clinicopathological variables or mutation status was examined using the χ2 test, continuity correction test, or Fisher's exact test. The rate of mature MIR 196A2 expression increase in mutation and no-mutation breast cancer tissue pairs was evaluated by Mann–Whitney U test. All statistical analyses were carried out with the SPSS 15.0 package (SPSS, Inc., Chicago, IL). All p-values were two tailed.
Results
SNP rs11614913 in MIR 196A2 undergoes frequent somatic mutations in breast cancer
We determined the genotypes of SNP rs11614913 in breast cancers; 114 tumor–normal (T–N) tissue pairs were available from the Cancer Hospital of Tianjin Medical University. The data showed that the genotype distribution of the functional variant was different in tumor tissues from that in the matched normal tissues (Table 1). The genotype frequencies of both TT and CC homozygous were increased 7.0% and 2.6% in tumor tissues, respectively, and the genotype frequency of CT heterozygous was decreased 9.6% in tumor tissues. An interesting observation was that the functional variant underwent somatic mutations in tumor tissue. Of the 114 T–N pairs, seven cases mutated from CT in the germ line to TT in the tumor, six cases mutated from CT to CC, two cases mutated from CC to CT, and one CC becomes TT (Fig. 1). We then calculated a parameter value (t) that might represent the resistance to the mutation susceptibility for the different genotype individuals. From Table 1, the ratio of the TT homozygous number in tumor tissue to the TT homozygous number in matched normal tissue is 33:25; the ratio of the CC homozygous number in tumor tissue to the CC homozygous number in matched normal tissue is 31:28, and the ratio of the CT heterozygous number in tumor tissue to the CT heterozygous number in matched normal tissue is 50:61. Here, if we set t (TT) as 1, then we get t (CC) as 0.84 and t (CT) as 0.62. Thus, the data indicated that the TT homozygous state appears to be resistant from mutation, while the CC homozygous and CT heterozygous states are more prone to mutation.
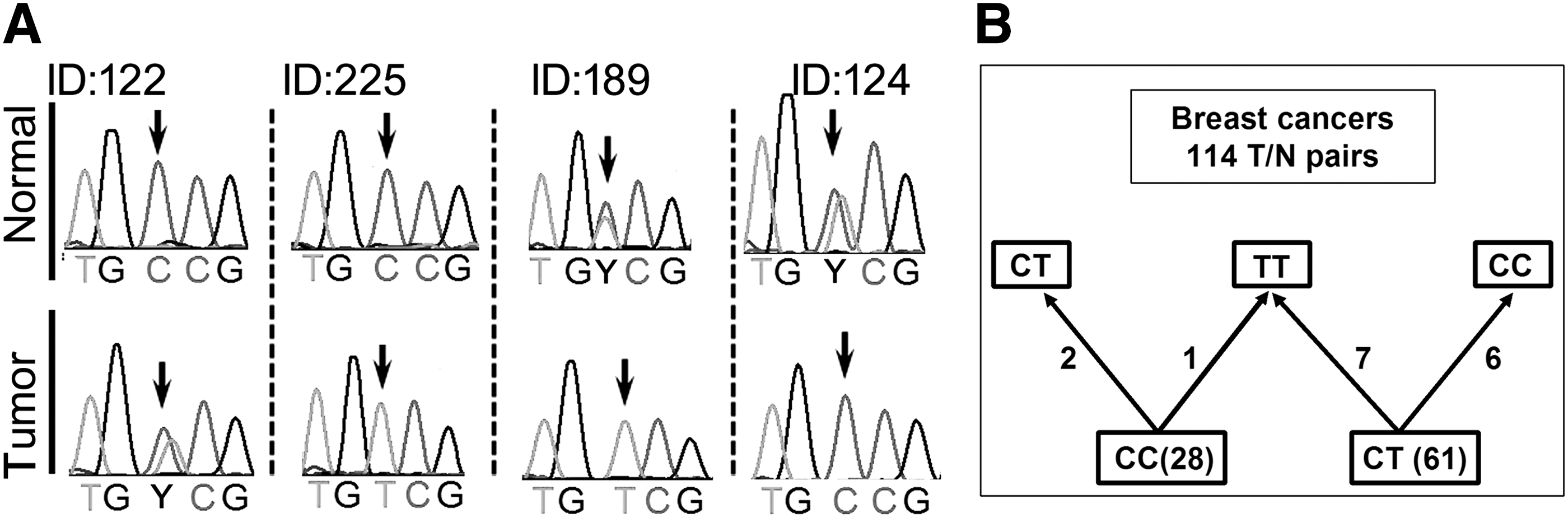
Somatic mutations in T–N tissue pairs.
The independent mutation rates of C→T and T→C are 0.094 and 0.054, respectively. Parameter value (t) represents the resistance to the mutation susceptibility for the different genotype individuals.
SNP, single-nucleotide polymorphism.
SNP rs11614913 was not associated with the pathological variables of breast cancer
We first correlated the mutation states with pathological variables of breast cancer; the result suggested that the mutation state was not associated with any pathological variables (data not shown). We then examined the association between the genotypes and the pathological variables. The CT and CC genotypes are more prone to mutation than the TT genotype, so we combined the CC and CT genotypes together and did the association analysis, but the result showed that genotype was also not associated with pathological variables (Table 2).
p-Values were determined using the χ2 test.
Expression of mature MIR 196A2 is increased in breast cancer
We analyzed the expression of mature MIR 196A2 in both cell lines and primary tumors from breast cancer. Compared to immortalized noncancerous mammary epithelial cell lines MCF10A and 184A1, most breast cancer cell lines examined, including BT-549, HCC1806, Hs578T, SW527, T-47D, and ZR-75–1, expressed higher levels of MIR 196A2. ZR-75–1 had the highest level, while MDA-MB-231 had no detectable expression (Fig. 2).
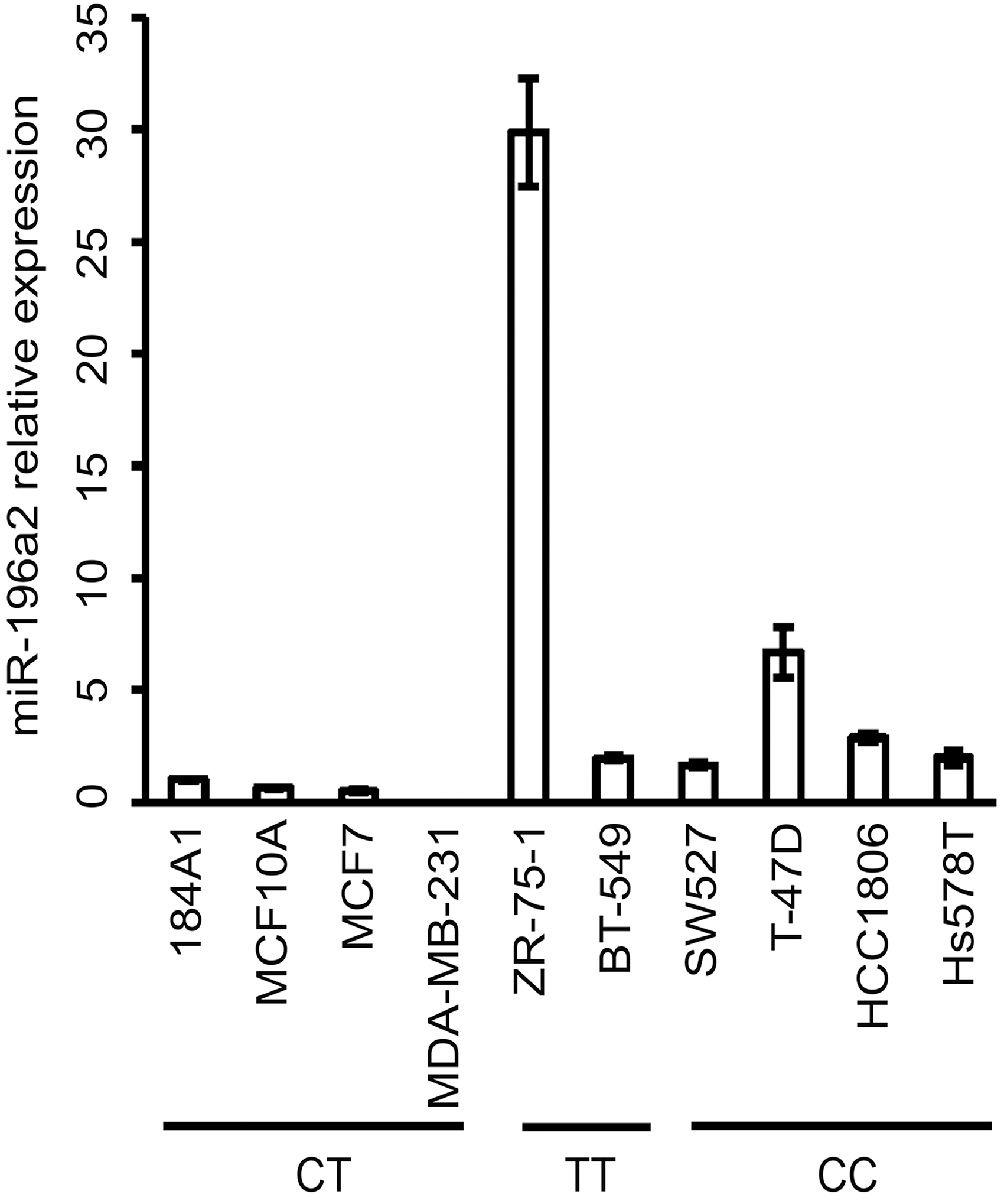
Expression of mature MIR 196A2 in nontumorigenic breast epithelial cell lines and breast cancer cell lines.
Forty-three paired normal and breast cancer tissues were used for miRNA expression analysis, including 10 cases with the CC genotype, 10 with the CT genotype, 10 with the TT genotype, and 13 cases that had somatic mutations in rs11614913. Among the total 43 paired tissues, 25/43 (58%) tumors had increased MIR 196A2 expression of at least 1.5-fold (T/N) compared to adjacent normal tissues (Supplementary Table S1; Supplementary Data are available online at
Analysis of 30 cases without mutation revealed that the average level of MIR 196A2 expression in tumor tissues was also significantly higher than that in adjacent normal tissues (t = 2.07, df = 29, p = 0.048). As shown in Table 3, the mean increased MIR 196A2 expression in the tumors with the CC genotype is 3.21 ± 1.00-fold; the mean increased MIR 196A2 expression in the tumors with the CT genotype is 2.76 ± 0.85-fold, and the mean increased MIR 196A2 expression in the tumors with the TT genotype is 1.19 ± 0.24-fold. However, expression analysis in the subset of tumors (n = 30, without SNP rs11614913 sequence alterations) revealed no significant difference in relative expression of MIR 196A2 by SNP rs11614913 genotype (ANOVA, p = 0.17).
p-Values were determined using the least significant difference test.
p-Value was determined using the F test with the general linear model.
SE, standard error.
Mature MIR 196A2 expression level significantly correlates with both the occurrence of SNP rs11614913 mutation and the higher tumor grade in breast cancer
In the 13 cases that had somatic mutations, the amount of mature MIR 196A2 in tumors was 1.7- to 173.6-fold of that in adjacent normal tissues. While in the 30 cases without the rs11614913 sequence alteration, the expression of mature MIR 196A2 in tumors was 0.4- to 9.3-fold of that in adjacent normal tissues, and only 12 of 30 cases had increased MIR 196A2 expression of at least 1.5-fold (Supplementary Table S1). That is, the rate of mature MIR 196A2 expression increase in the 13 cases is significantly higher than that in the 30 cases, as determined by the Mann–Whitney U test (Z = −3.7, p = 0.0002) (Fig. 3). Moreover, the Fisher's exact test result indicated that the occurrence of rs11614913 mutation is significantly associated with the higher expression level of MIR 196A2 in the tumors (p = 0.0002) (Table 4).
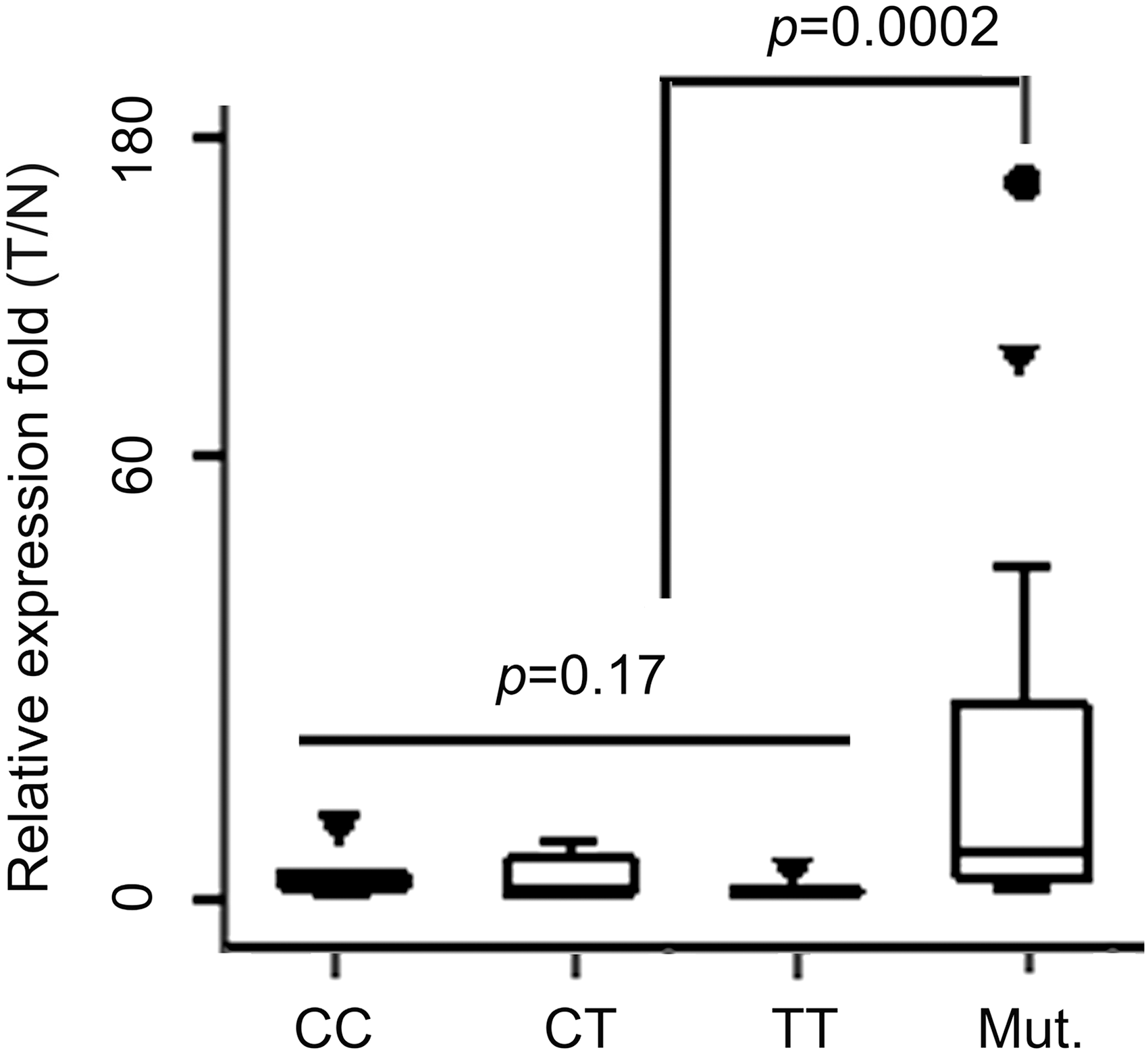
A comparison of mature MIR 196A2 expression between mutation and no-mutation breast cancer tissue pairs. The relative increased fold (T/N) of mature MIR 196A2 expression in the 13 cases that underwent rs11614913 somatic mutation compared to that in the 30 cases without sequence modifications detected using real-time reverse transcription–polymerase chain reaction.
p-Value for the remaining cases was calculated using the χ2 test.
p-Value for tumor grade was calculated using continuity correction test.
p-Value for mutation was calculated using Fisher's exact test.
The χ2 test and the continuity correction test were applied to evaluate the association between increased MIR 196A2 expression and clinicopathological variables of breast cancer, and a significant association was detected with the higher tumor grade as 15 of 17 (88.2%) grade III tumors and only 10 of 26 (38.5%) grade I + II tumors had an overexpression of MIR 196A2, with a T:N ratio of at least 1.5 (continuity correction test, p = 0.004) (Table 4).
Discussion
SNPs in miRNA genes and the genes of their target sites may function as regulatory factors to alter phenotypes and disease susceptibility. Somatic mutations of SNPs located in miRNAs have been demonstrated to associate with diseases, including cancer, likely by affecting miRNA maturation and target selection. Germline mutations or rare SNPs in the primary transcripts of has-mir-15a and has-mir-16–1 have been linked to familial chronic lymphocytic leukemia and familial breast cancer (Calin et al., 2005; Raveche et al., 2007). A common SNP in pre-miR-146a decreases mature miR expression and predisposes to papillary thyroid carcinoma (Jazdzewski et al., 2008). A single mutation in pre-miR-155 leads to a shift in strand selection and thus results in a global expression alteration (Lee et al., 2011). A mutation in the seed region of human miR-96 was responsible for nonsyndromic progressive hearing loss (Mencia et al., 2009). In addition, a synonymous variant in immunity-related GTPase Family M (IRGM) alters a binding site for miR-196 and causes deregulation of IRGM-dependent xenophagy in Crohn's disease (Brest et al., 2011). MIR 196A2 was highly expressed in breast and pancreatic cancers, and the elevated expression was associated with significantly reduced survival for pancreatic cancer. In addition, a growing number of studies have suggested that C allele in the MIR 196A2 gene is emerging as a low-penetrance tumor susceptibility allele in the tumorigenesis. However, the results are inconclusive.
In the present study, we found that SNP rs11614913 in MIR 196A2 undergoes frequent somatic mutations in breast cancer. Analysis of the mutations revealed that the CT heterozygous and the CC homozygous states are more prone to mutation, while the TT homozygous state appears to be resistant. The data indicated that the CC or CT genotypes have a higher risk of gaining somatic alteration during malignant transformation of breast cancer. The relatively frequent (14%) occurrence of tumor-specific alteration of SNP rs11614913 provided additional support for the conclusion that MIR 196A2 contributes to the development and/or progression of breast cancer. We further calculated the independent mutation rate, and the independent mutation rate of C→T is 0.094, while that of T→C is 0.054. The results did not support that MIR 196A2 C allele is a low-penetrant risk factor for breast cancer development. But somehow, the MIR 196A2 C allele might result in loosing the chromatin to control the availability of genomic loci to transcriptional or other regulation. As C→T is in the majority of mutation cases, and somatic mutation was statistically significantly associated with increased mature MIR 196A2 expression in the tumors.
It has been shown that MIR 196A2 was highly expressed in pancreatic (Bloomston et al., 2007) and breast (Iorio et al., 2005; Hui et al., 2009) cancers compared to normal tissues. Consistent with these studies, we found an elevated expression of mature MIR 196A2 in breast cancers. We also found no significant difference in relative expression of MIR 196A2 by SNP rs11614913 genotype. Hu et al. (2008) observed significantly higher expression of miR-196a in nonsmall cell lung tumor samples with the CC genotype (n = 6) compared to CT and TT individuals (n = 17). Hoffman et al. (2009) showed increased expression of mature miR-196a in breast cancer cells transfected with pre-miR-196a-C compared to cells transfected with miR-196a-T relative to empty vector control. Christensen et al. (2010) showed no significant difference in relative expression of miR-196a by rs11614913 genotype in the head and neck squamous cell carcinoma. Relative to previous work, our expression results were from a number of samples (n [CC] = 10, n [CT] = 10, and n [TT] = 10) without sequence alteration of the SNP rs11614913. In addition, the data showed no significant difference in relative expression of MIR 196A2 by SNP rs11614913 genotype. If SNPs located in the miRNA genes directly affect the amount of mature miRNA, then the functional relationship between these SNPs and disease susceptibility or prognosis may seem obvious, and altered miRNA expression leads to altered regulation of target mRNAs important in tumorigenesis. In addition, variation in the mature miRNA sequence itself could result in differential regulation of target mRNAs. In the present study, we found that the occurrence of SNP rs11614913 mutation was statistically significantly associated with increased mature MIR 196A2 expression. Therefore, the reason that caused rs11614913 mutation might be responsible for the dysregulation of mature MIR 196A2 expression. In addition, mature MIR 196A2 expression level significantly correlates with the higher tumor grade in breast cancer.
In summary, we identified that a functional variant rs11614913 in MIR 196A2 was frequently mutated in breast cancers. In addition, the CT heterozygous and the CC homozygous states were more prone to mutation, while the TT homozygous state appeared to be resistant. The expression level of mature MIR 196A2 was highly elevated in breast cancers. Furthermore, the mutation state of SNP rs11614913 was statistically significantly associated with the higher expression level of mature MIR 196A2 in the tumors. In addition, mature MIR 196A2 expression level was significantly associated with the higher tumor grade. These findings provided additional support for the conclusion that MIR 196A2 contributes to the development and/or progression of breast cancer. However, the results did not support that MIR 196A2 C allele is a low-penetrant risk factor for breast cancer development. However, it indicated that an unknown mechanism, which might promote rs11614913 mutation and might be responsible for the dysregulation of mature MIR 196A2 expression, remains to be determined.
Footnotes
Acknowledgments
This work was supported by grants from the National Nature Science Foundation of China (grant nos. 81470118, 81272219, 31171250, and 30930038), the Nature Science Foundation of Hebei Province (grant No. H2012401036), the Open Fund of State Key Laboratory of Medicinal Chemical Biology (Nankai University), and the Undergraduate Science and Technology Innovation Fund (Tianjin Medical University).
Disclosure Statement
No competing financial interests exist.
References
Supplementary Material
Please find the following supplemental material available below.
For Open Access articles published under a Creative Commons License, all supplemental material carries the same license as the article it is associated with.
For non-Open Access articles published, all supplemental material carries a non-exclusive license, and permission requests for re-use of supplemental material or any part of supplemental material shall be sent directly to the copyright owner as specified in the copyright notice associated with the article.