
Abstract
Ulcerative colitis (UC) is a long-time inflammatory condition arising from aberrant immune activation in the colon and the rectum. Interleukin (IL)-35 plays critical roles in autoimmune disorders. In this study, we explored the pathways of IL-35 in affecting UC. First, peripheral blood mononuclear cells (PBMCs) from UC patients were obtained. Pretreating PBMCs with IL-35 resulted in significantly elevated IL-10 production from whole PBMCs as well as B cells, whereas pretreating PBMCs with IL-12 or IL-27 did not demonstrate a similar effect. IL-35 suppressed the proliferation of CD4+CD25− conventional T cells, CD4+CD25+ regulatory T (Treg) cells, and CD8+ T cells, but did not inhibit the proliferation of B cells. IL-35-mediated IL-10 secretion in B cells did not require the presence of Treg cells. After treatment with IL-35, B cells from UC patients presented significantly enhanced regulatory function, characterized by inhibiting cell proliferation and interferon (IFN)-γ, IL-17, and tumor necrosis factor (TNF)-α secretion from autologous CD4+CD25− T cells and CD8+ T cells, which was dependent on IL-10 signaling. However, IL-35-treatment did not demonstrate an effect on regulating IL-5 and IL-13 responses. These discoveries identified a Th1, Th17, and CD8+ T cell-targeting role of IL-35 in UC patients. Next, we examined the IL-35 expression in the intestinal mucosal in UC patients. Data showed that both noninflamed and inflamed tissues from UC patients presented significantly lower IL-35 secretion compared to healthy control tissues, which was associated with suppressed p35 transcription. UC patients with higher IL-35 also presented higher IL-10 secretion in gut mucosa. Together, our study identified that IL-35 could mediate anti-inflammatory function through promoting regulatory B cell functions, but this effect was suppressed in UC patients.
Introduction
U
Central to all UC symptoms is the pathogenic inflammation mediated by the immune system. The inflamed areas of the gut in UC patients express high levels of chemoattractants and adhesion molecules, which lead to local infiltration and accumulation of T cells, elevated cell cycling, and resistance against apoptosis (Sturm et al., 2004; Van Assche and Rutgeerts, 2005; Mudter and Neurath, 2007). Adoptive transfer of naive CD4+CD45RBhiCD25− conventional T cells initiated inflammatory bowel disease (IBD) in Rag1−/− mice (Izcue et al., 2006; Collison et al., 2010), while blocking T cell inflammation attenuated mucosal inflammation in murine colitis models (Monteleone and Caprioli, 2010). Therefore, an effective UC treatment strategy could be targeting the immune dysregulation, suppressing pathogenic inflammation, and restoring tissue integrity and mucosal homeostasis.
Regulatory B (Breg) cells exert important inhibitory functions in the healthy immune system. Breg cells are characterized by the production of interleukin (IL)-10 (Rosser and Mauri, 2015), a pleiotropic cytokine that is essential in limiting ongoing inflammation, protecting tissue integrity, and facilitating injury healing (Ouyang et al., 2011). In mice, IL-10-secreting B cells were required to prevent the development of autoimmunity (Mizoguchi et al., 1997; Fillatreau et al., 2002; Mauri et al., 2003; Matsushita et al., 2008). Deficiency in B cell regulatory function was observed in multiple human autoimmune diseases, such as multiple sclerosis and systemic lupus erythematosus (Duddy et al., 2007; Blair et al., 2010). Interestingly, depletion of B cells with rituximab led to exacerbated UC symptoms in some patients (Goetz et al., 2007; El Fassi et al., 2008). Moreover, a downregulation of Breg cell subsets, including CD24hiCD38hi B cells and CD5+ B cells, was identified in UC patients (Wang et al., 2016). These results suggested that the B cell regulatory activity was also dysregulated in UC patients. An urgent need to identify the UC-specific factors promoting or inhibiting Breg cells is present.
Recently, IL-35 as a key inducer of B cell regulatory activity is being recognized (Tedder and Leonard, 2014; Rosser and Mauri, 2015). IL-35 is a heterodimer composed of the IL-12 subunit p35 and the IL-27 subunit Epstein-Barr virus-induced gene 3 (EBI3). Mice lacking either p35 or EBI3 in B cells presented a more severe form of experimental autoimmune encephalomyelitis (EAE) as well as enhanced anti-Salmonella immunity (Shen et al., 2014). IL-35-stimulated B cells produced IL-10 and IL-35, and were capable of uveitis inhibition on adoptive transfer (Wang et al., 2014). The expression of IL-35 in UC and its roles in modulating immune responses are therefore of great interest for the improved understanding of the immune responses in UC and development of novel treatment strategies. In this study, we examined the role of IL-35 in regulatory T (Treg) cells and Breg cells in UC.
Materials and Methods
Subjects
Peripheral blood was investigated in 10 (6 female and 4 male subjects between 49 and 71 years) UC patients with active disease. Diagnosis was made in Shandong Provincial Hospital according to standard endoscopic, histological, and radiological criteria (Silverberg et al., 2005). All patients were untreated at the time of sample collection and UC patients using immunomodulators or corticosteroids were excluded. Biopsy specimens were obtained from macroscopically noninflamed and inflamed areas of UC patients during ileocolonoscopy. Healthy control biopsy specimens were obtained from subjects who received colonoscopy during colorectal cancer screening, with a negative result. The healthy controls who provided biopsies were age- and gender-matched with UC patients, with four females and six males between 55 and 74 years. Clinical information was analyzed by DICAT (Vancouver, Canada). This study was performed with protocols approved by the ethics board of Shandong Provincial Hospital Affiliated to Shandong University. Written informed consent was provided by every subject.
Harvesting mononuclear cells
Peripheral blood mononuclear cells (PBMCs) were harvested from fresh blood by Ficoll gradient centrifugation and then stored at −80°C in 10% dimethyl sulfoxide (DMSO) and 90% fetal bovine serum. Intestinal mononuclear cells (IMCs) were harvested using enzymatic digestion (Maul et al., 2005). Briefly, fresh biopsies were washed and incubated in sterile phosphate-buffered saline (PBS) +1 mM EDTA for 30 min at 4°C, washed again in pure PBS, and then digested with 0.1 mg/mL collagenase, 200 μg/mL hyaluronidase, and 100 μg/mL DNAse (Sigma-Aldrich) in Hank's Balanced Salt Solution for 1 h in a 37°C shaking water bath. The resulting cell suspension was pushed through a 70 μm cell strainer (BD) and centrifuged with Ficoll to obtain mononuclear cells.
Cell subtype isolation
CD8+ T cells and B cells were isolated from PBMCs using the Human CD8+ T cell enrichment kit and the Human B cell enrichment kit (Stemcell), respectively, which employed negative magnetic sorting strategy. CD4+CD25− were isolated using the Human CD4+ T cell enrichment kit (BioLegend) with an additional anti-human CD25 mAb (clone BC96; BioLegend) in the antibody cocktail to remove the CD4+CD25+ Treg cells. CD4+CD25+ Treg cells were positively sorted by using the Human CD4+CD25+ T cell isolation kit (Stemcell).
Cytokines, antibodies, and other reagents
The following recombinant human (rh) proteins were used as exogenous cytokines in experiments: carrier-free rhIL-12 (catalogue No. 219-IL/CF; R&D Systems), carrier-free rhIL-27 (catalogue No. 589204; BioLegend), and carrier-free rhIL-35 (catalogue No. 8608-IL; R&D Systems). Bovine serum albumin (BSA; low endotoxin, fatty acid free, sterile filtered) was purchased from Sigma-Aldrich (catalogue No. A1595). IL-10 blocking mAb (clone JES3-9D7) and IL-10 receptor-α (IL-10Rα) blocking mAb (catalogue No. MAB274) were purchased from BioLegend and R&D Systems, respectively. SEB was purchased from Sigma-Aldrich (catalogue No. S4881).
Cytokine detection
Secreted IL-10 was detected using Human IL-10 enzyme-linked immunosorbent assay (ELISA) kit (Thermo Fisher Scientific). Secreted IL-35 was detected using anti-EBI3 (catalogue No. ab83896; Abcam) capture antibodies and biotinylated anti-p35 (clone IL-12-III; MabTech) detection antibody, together with avidin-HRP and TMB solution. To detect transcript levels, mRNA was first extracted using the RNeasy Mini kit (Qiagen) and transcribed to cDNA using the SuperScript III kit (Thermo Fisher Scientific). The IL-10, p35, and EBI3 transcripts were then detected using the SYBR Green and Applied Biosystems 7500 Fast Real-Time PCR System, normalized to GAPDH. Secretion of interferon (IFN)-γ, IL-5, IL-13, IL-17, and tumor necrosis factor (TNF)-α was detected using Luminex following manufacturer's instructions (EMD Millipore).
Cell proliferation
Proliferation was measured by pulsing cells with 0.5 μCi per 10 μL of 3H-thymidine in each well, as described (Oh et al., 2011). All conditions were triplicated for the calculation of the mean value.
Statistics
Comparisons between two datasets were performed using Welch's t-test (parametric) or Mann-Whitney test (nonparametric). Comparisons between multiple datasets were performed using one-way or two-way analyses of variance (ANOVAs), followed by Bonferroni's (parametric) or Dunn's (nonparametric) post-test. All tests were performed by PRISM 5 software.
Results
In B cells, rh IL-35 induced IL-10 secretion, but did not suppress cell proliferation
A previous study identified an impairment of Breg cell subsets in UC, characterized by reduced frequencies of CD24highCD38high B cells and CD5+ B cells, and reduced serum IL-10 level (Wang et al., 2016). It is yet unclear whether Breg cell functionalities in UC patients could be revived by stimulation with regulatory cytokines. We first investigated a strategy using IL-35, a cytokine shown to have crucial roles in the development of Breg cells in mice (Shen et al., 2014; Wang et al., 2014). Since IL-35 shares subunits with IL-12 and IL-27, those two cytokines were used as controls, together with a protein control BSA. PBMCs were harvested from UC patients and were cultured with exogenous BSA, rhIL-12, rhIL-27, and rhIL-35. After 3 days, whole PBMCs or purified B cells isolated from whole PBMCs were stimulated with SEB for 24 h. The IL-10 secretion of whole PBMCs was examined by ELISA on the supernatants harvested daily. In the SEB-stimulated whole PBMCs, exogenous rhIL-35 significantly increased the secretion of IL-10 (Fig. 1a).
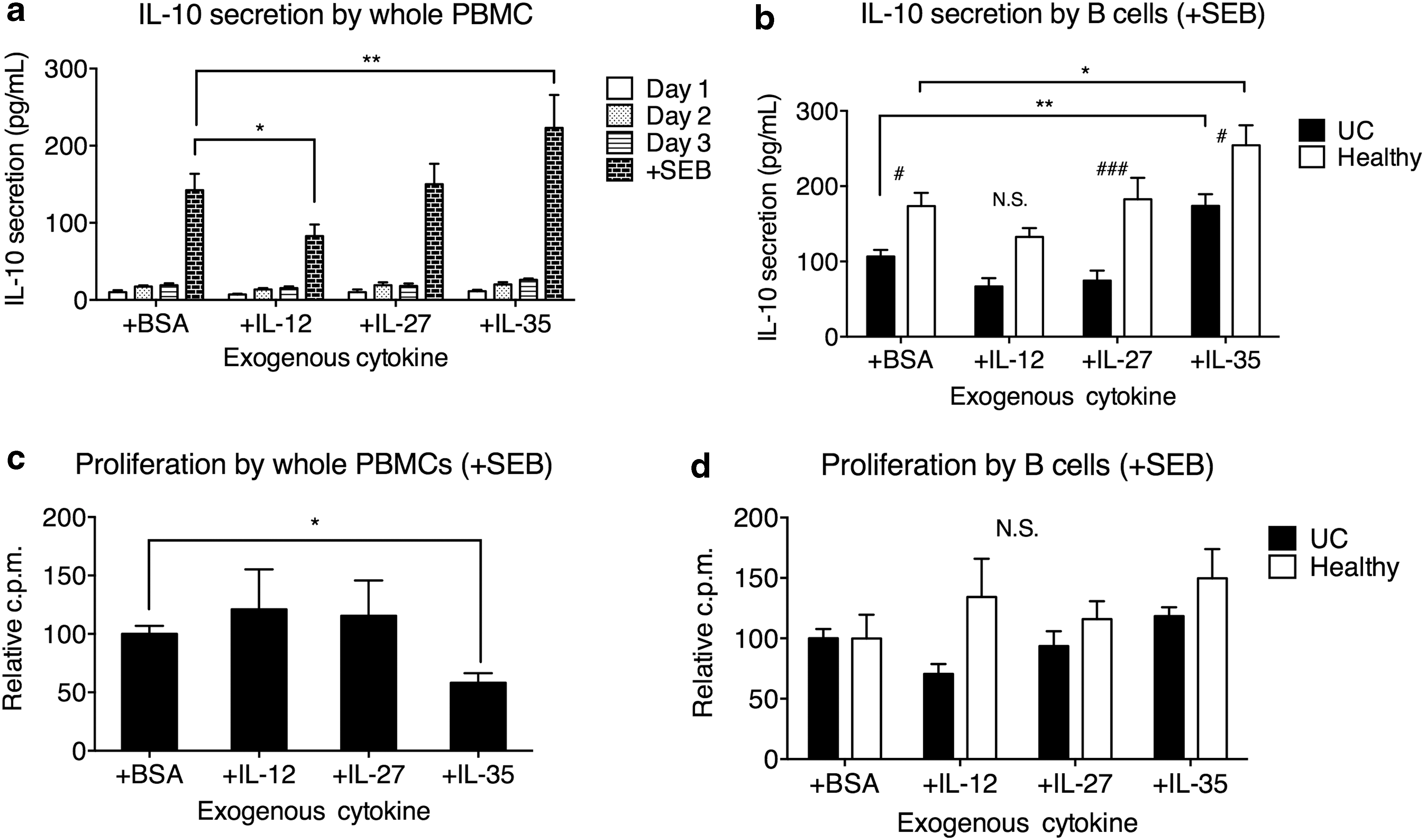
Exogenous IL-35 induced IL-10 production in PBMCs and B cells, and suppressed PBMC proliferation. PBMCs were incubated in the presence of 5 μg/mL BSA, IL-12, IL-27, or IL-35 for 3 days at 2 × 105 cells/mL, and then for 24 h with 2 μg/mL SEB. One hundred microliters of supernatant was obtained daily and replaced with fresh media with the same formulation. For B cell analyses, B cells were isolated from whole PBMCs and cultured alone at 2 × 105 cells/mL during SEB stimulation.
IL-35 by itself in the absence of stimulation did not lead to IL-10 production. We then examined the IL-10 secretion by SEB-stimulated purified B cells from UC patients and healthy controls. We found that IL-35 significantly increased the B cell IL-10 secretion in both UC patients and healthy controls (Fig. 1b). Consistent with previous study, healthy control B cells presented elevated IL-10 secretion than UC patients, with or without additional IL-35. The control cytokines IL-12 and IL-27 did not significantly increase the production of IL-10 from whole PBMCs or purified B cells.
Previously, it was demonstrated that exogenous IL-35 suppressed lymphocyte proliferation (Wang et al., 2014). In this study, we examined the 3H-thymidine incorporation by whole PBMCs or purified B cells following 3 days of treatment with BSA, IL-12, IL-27, and IL-35, and 24 h of SEB stimulation. After SEB stimulation, IL-35 treatment significantly reduced the proliferation of whole PBMCs, while IL-12 and IL-27 did not exert a significant effect (Fig. 1c). No cytokine significantly altered the proliferation of B cells in UC patients or healthy controls (Fig. 1d).
To examine the minimum concentration required to enhance the IL-10 secretion by B cells from UC patients, we added IL-35 at various concentrations. The B cells were then stimulated with SEB as described above. We found that IL-35 at 5 ng/mL and 5 μg/mL could significantly upregulate B cell IL-10 secretion (Fig. 2a). No significant change in B cell proliferation was seen at any IL-35 concentration (Fig. 2b).
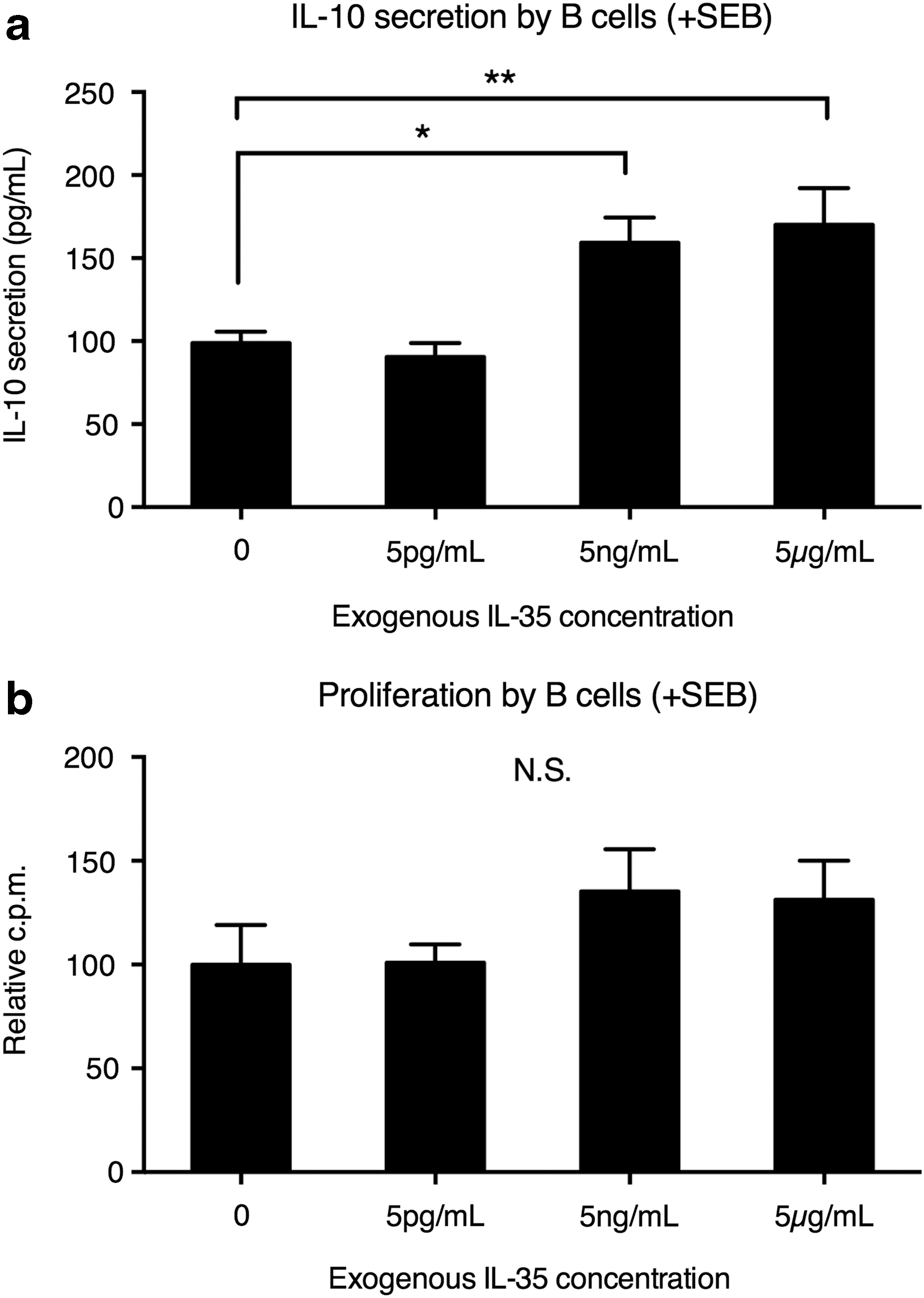
IL-10 secretion and proliferation by UC B cells under various concentrations of IL-35. PBMCs from UC patients were incubated in the presence of 0, 5 pg/mL, 5 ng/mL, or 5 μg/mL IL-35 for 3 days at 2 × 105 cells/mL. The B cells were then isolated and stimulated for 24 h with 2 μg/mL SEB.
Regulatory actions of IL-35 on B cells did not require CD4+CD25+ Treg cells
In T cells, IL-35 exerted regulatory function by suppressing T cell proliferation and converting conventional T cells to IL-35-producing cells, with concurrent increase in CD25 expression (Collison et al., 2010). We asked whether IL-35 induced IL-10 production of B cell directly or indirectly through Treg cells. We found that CD4+CD25+ Treg cell depletion did not significantly alter the IL-35-induced IL-10 secretion (Fig. 3a). IL-35 treatment reduced the proliferation of CD4+CD25+ Treg cells, CD4+CD25− T cells, and CD8+ T cells, but did not significantly change the proliferation of B cells (Fig. 3b). Together, these results demonstrated that the induction of B cell IL-10 by IL-35 did not require the presence of CD4+CD25+ Treg cells.

IL-35 did not require Treg cells to exert regulatory functions on B cells.
IL-35-induced Breg cells inhibited CD4+CD25− T cell inflammation
UC was traditionally thought to be mediated by aberrant Th2-type inflammation, primarily due to the observations that Th2 cytokines IL-5 and IL-13 were overrepresented in mucosal tissues with UC (Fuss et al., 1996, 2004; Strober and Fuss, 2011). In recent years, elevated IL-17 under IL-23 induction was observed in UC patients and was associated with disease severity (Kobayashi et al., 2008). Breg cells were previously observed to favor the development and maintenance of Treg cells over the Th1 or Th17 cells (Matsushita et al., 2008; Carter et al., 2011), and were shown to directly inhibit the degranulation and cytokine production of activated CD4+ T cells and CD8+ T cells (Liu et al., 2014). UC patients presented downregulated Breg cell frequency and activity (Wang et al., 2016). It remained unclear whether Breg cells could exert inhibitory action on Th2 cells and Th17 cells in UC patients, and whether IL-35 could rescue the Breg cell deficiency in UC patients.
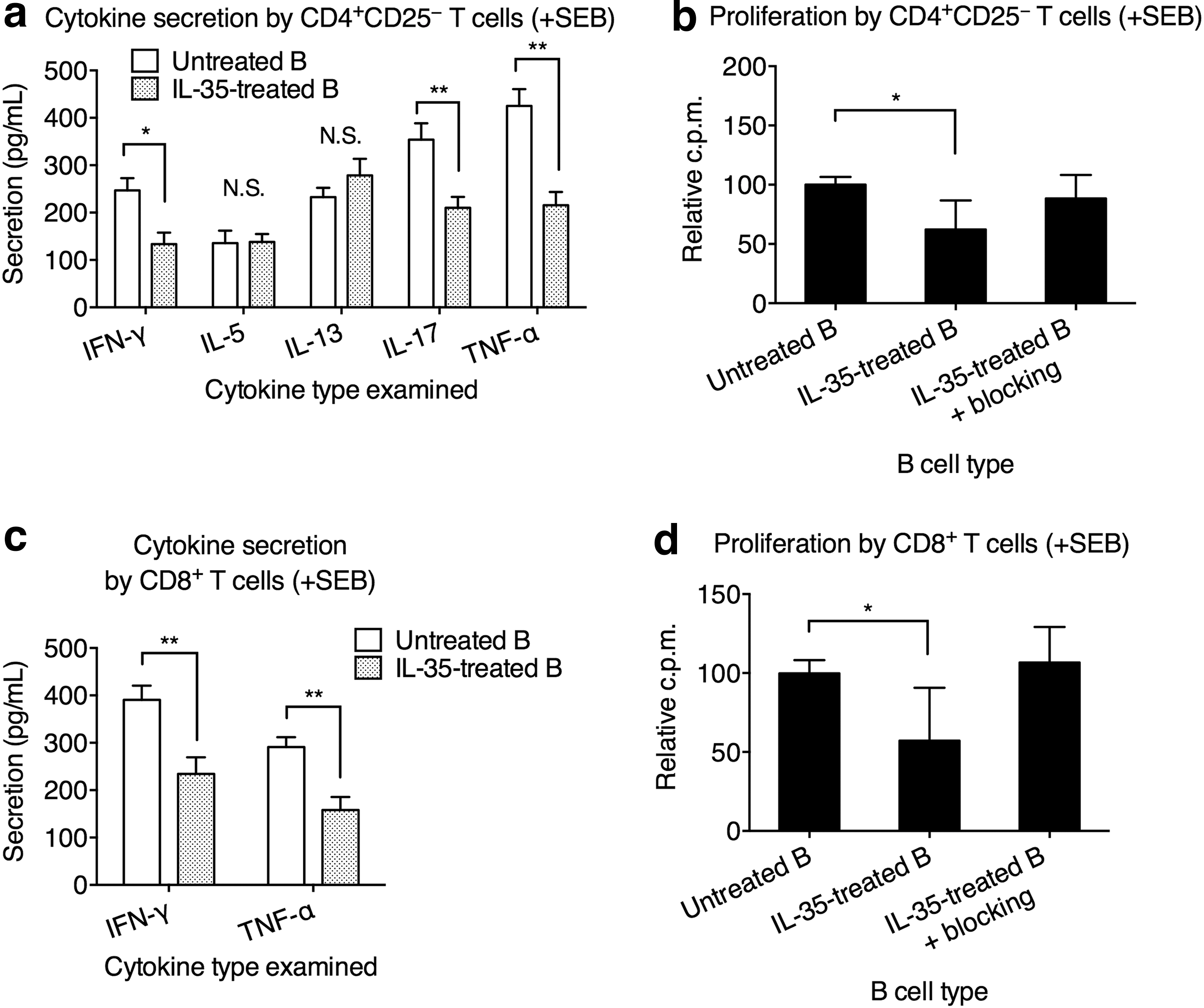
To investigate this, we cocultured CD4+CD25− T cells with autologous B cells (untreated B) or autologous B cells pretreated with IL-35 for 3 days (IL-35-treated B). The CD4+CD25+ Treg cells were not included because first, they did not participate in IL-35-mediated regulation of B cell, and second, we wanted to avoid confounding effects of Treg cells. After coculture, the T cells were isolated and stimulated with 2 μg/mL SEB. The cytokine secretion was measured after 24 h. We found that compared to untreated B cells, IL-35-treated B cells significantly reduced the secretion of IFN-γ, IL-17, and TNF-α by T cells (Fig. 4a). The secretion of IL-5 and IL-13 from T cells was unaffected whether untreated B cells or IL-35-treated B cells were used. Blocking either IL-10 or IL-10R partially reduced the IL-35-treated B cell-mediated inhibition of IL-17 and/or TNF-α production from CD4+CD25− T cells, while blocking both IL-10 and IL-10R completely removed the inhibition of IFN-γ, IL-17, and TNF-α production from CD4+CD25− T cells (Fig. 4b). IL-35-treated B cells also suppressed the proliferation of CD4+CD25− T cells, which could be reverted by blocking IL-10 and IL-10R (Fig. 4c). These results strongly suggested that IL-35-treated B cells mediated inhibitory effects on CD4+CD25− T cells through elevating IL-10. Indeed, we found that IL-35 treatment significantly elevated the IL-10 transcription and secretion in B cells from UC patients (Fig. 4d).
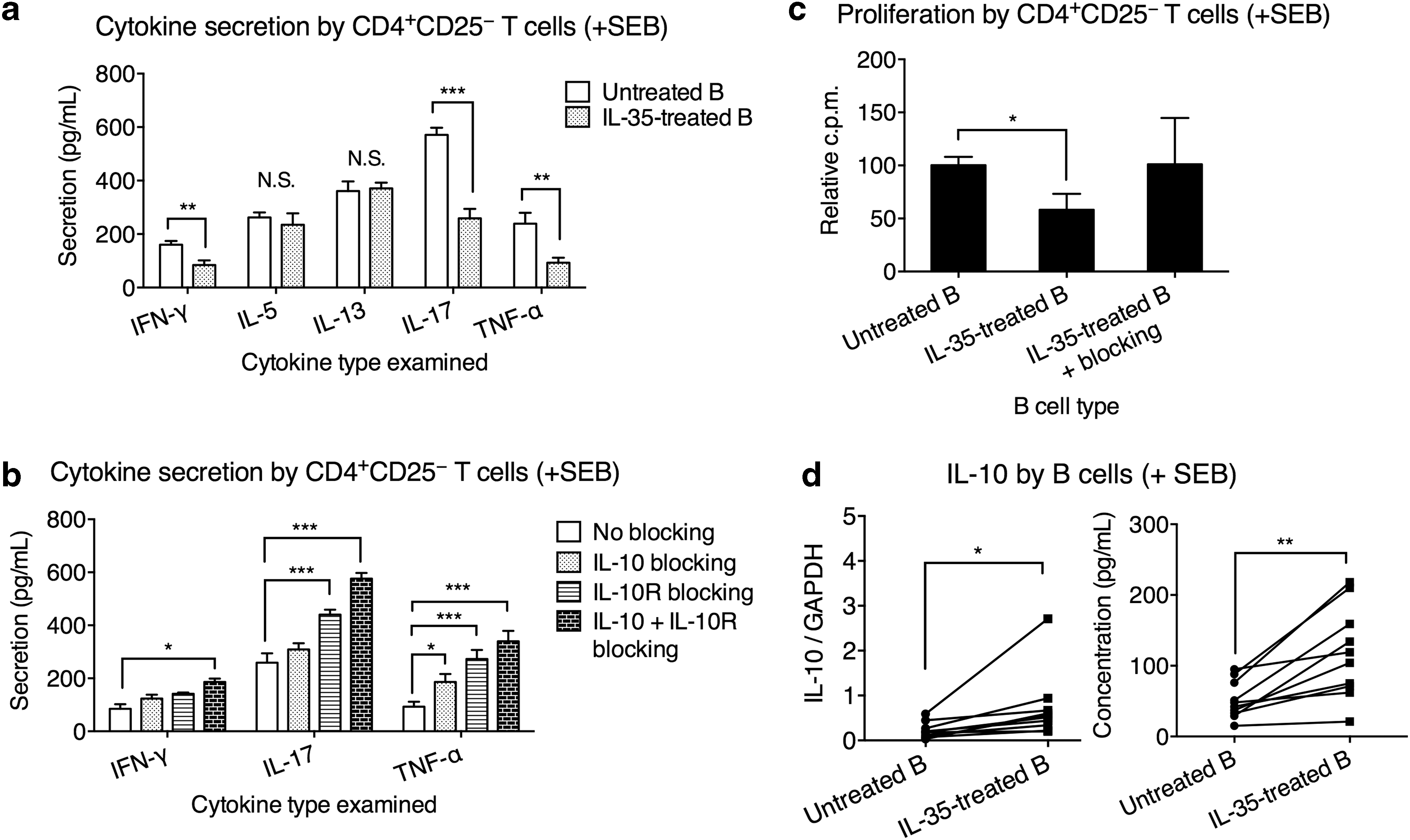
IL-35-treated B cells from UC patients inhibited CD4+CD25− T cell inflammation through IL-10 signaling. IL-35-treated B cells were obtained from UC patients' PBMCs that were treated with 5 μg/mL IL-35 for 3 days. Untreated B cells were obtained from UC patients' PBMCs that were treated with 5 μg/mL BSA control. Freshly isolated CD4+CD25− T cells were cocultured with IL-35-treated B cells or untreated B cells at 1:1 ratio for 3 days with SEB stimulation. The T cells were then separated and cultured alone for an additional 24 h with SEB, after which the cytokine secretion and proliferation were measured.
IL-35-treated B cells gained the ability to suppress CD8+ T cell inflammation
It was shown that CD8+ T cell gene expression profile was associated with the clinical outcome of UC and Crohn's disease (Lee et al., 2011). Patients with high CD8+ T cell expression of genes downstream of T cell receptor signaling and IL-2/IL-7 signaling required more treatment escalations, such as immunomodulators and surgery, and were still more likely to experience a refractory and chronically active disease. Therefore, we investigated whether IL-35-treated B cells could mediate the regulation of CD8+ T cells in UC patients. The previous experiments were repeated using purified CD8+ T cells to replace CD4+CD25− T cells. We found that compared to untreated B cells from UC, the IL-35-treated B cells potently suppressed the production of IFN-γ and TNF-α from autologous CD8+ T cells (Fig. 5a). By blocking IL-10 and/or IL-10R, the IL-35-treated B cell-mediated inhibition is alleviated (Fig. 5b). IL-35-treated B cells also suppressed the proliferation of CD8+ T cells, an effect inhibited by blocking IL-10 and IL-10R (Fig. 5c).
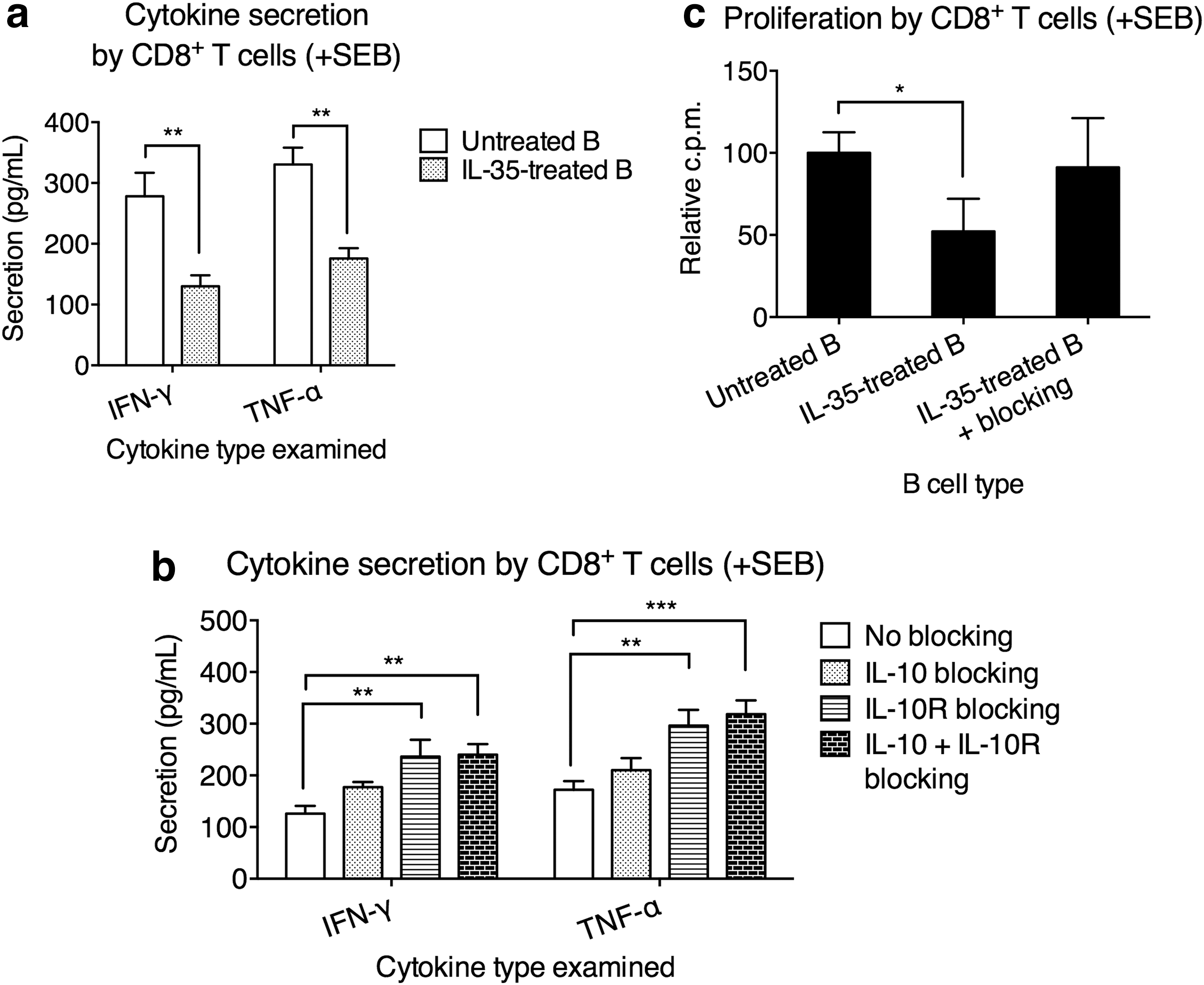
IL-35-treated B cells from UC patients inhibited CD8+ T cell inflammation through IL-10 signaling.
IL-35-treated B cells from healthy controls demonstrated similarly enhanced regulatory function toward autologous CD4+CD25− and CD8+ T cells
To examine whether the enhancement in CD4+CD25− and CD8+ T cell suppression by IL-35-treated B cells was restricted to UC patients, we repeated these experiments using T cells and B cells harvested from healthy controls. We found that IL-35-treated B cells from healthy controls significantly suppressed IFN-γ, IL-17, and TNF-α secretion from CD4+CD25− T cells (Fig. 6b), as well as IFN-γ and TNF-α production from CD8+ T cells (Fig. 6c). Furthermore, the proliferation CD4+CD25− T cells and CD8+ T cells was suppressed by IL-35-treated B cells, a function blocked by IL-10 and IL-10R monoclonal antibodies (Fig. 6b, d).
IL-35-treated B cells from healthy controls inhibited CD4+CD25- and CD8+ T cell inflammation. IL-35-treated or untreated B cells were obtained from healthy control PBMCs that were treated with 5 μg/mL IL-35 or 5 μg/mL BSA for 3 days, respectively. Freshly isolated autologous CD4+CD25− or CD8+ T cells were cocultured with IL-35-treated B cells or untreated B cells at 1:1 ratio, for 3 days with SEB stimulation. The T cells were then separated and cultured alone for an additional 24 h with SEB, after which the cytokine secretion and proliferation were measured.
IL-35 secretion is impaired in intestinal biopsies of UC patients due to impaired p35 production
Together, the previous experiments have demonstrated that compared to B cells directly from UC patients, IL-35-treated B cells were potent suppressors of Th1, Th17, and CD8+ T cell inflammation. We therefore asked the status of IL-35 secretion in UC patients. Intestinal biopsy specimens from macroscopically noninflamed and inflamed areas of UC patients and the noninflamed area of healthy controls were obtained, from which IMCs were harvested. Using a sandwich ELISA with EBI3-specific antibodies as capture antibody and anti-human p35 mAb as detection antibody, in a preliminary experiment in which titrated concentrations of rh IL-12, IL-27, IL-35, and BSA were tested, we found that this ELISA strategy effectively detected IL-35, but not other cytokines (Fig. 7a).
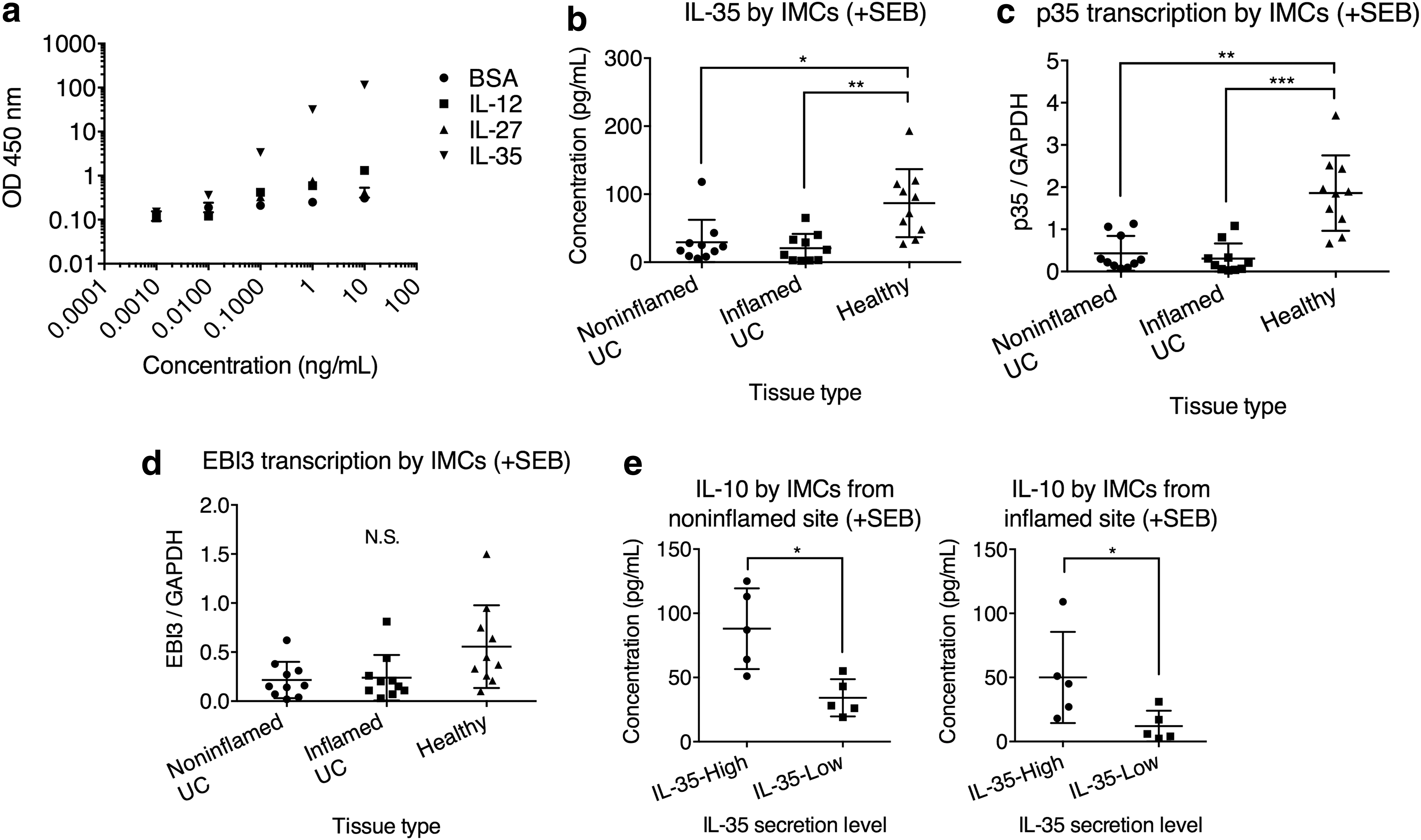
IL-35 production was lower in UC IMCs and was correlated with IL-10 production. IMCs from noninflamed and inflamed UC tissues, as well as IMCs from healthy individuals, were stimulated with 2 μg/mL SEB for 24 h, after which cells and supernatant were harvested for cytokine transcription and secretion, respectively.
We then examined the IL-35 secretion by IMCs. IMCs from both noninflamed and inflamed tissues from UC patients presented significantly lower IL-35 secretion than IMCs from healthy controls (Fig. 7b). Between noninflamed and inflamed tissues from UC, there was no statistically significant difference. We then examined the transcription level of the two subunits of IL-35, p35 and EBI3. Interestingly, we found that the p35 transcription was significantly lower in IMCs from the noninflamed and inflamed tissues from UC patients than that from healthy controls (Fig. 7c), while the EBI3 transcription was not significantly different among the three groups (Fig. 7d), indicating that low IL-35 level in IMCs was possibly due to a suppression of IL-35 subunit p35. We also observed that in UC patients with high IL-35, the secreted IL-10 level by IMCs was also higher compared to those with low IL-35 (Fig. 7e).
Discussion
UC patients displayed a lack of Breg cells (Wang et al., 2016). Spontaneous UC was developed in TCRα-deficient mice and was further exacerbated in mice with additional B cell deficiency (TCRα-deficient Igμ-deficient) (Mizoguchi et al., 2000). IL-10-producing Breg cells (induced by IL-33) blocked the spontaneous development of mucosal inflammation in the gut of IL-10-deficient mice (Sattler et al., 2014). These studies together suggested that the pathogenesis of UC involved a deficiency of Breg cells, and treatment that could increase B cell regulatory function might serve as an effective option to treat UC. IL-35 is known to play an essential role in the development of Treg cells and Breg cells, with immunosuppressive activities (Collison et al., 2010; Wang et al., 2014). In mice, IL-35 converted B cells into IL-10-producing and IL-35-producing Breg cells, which suppressed multiple autoimmune disorders as well as inhibited anti-Salmonella immunity (Shen et al., 2014; Wang et al., 2014). Therefore, in this study, we investigated the role of IL-35 in regulating immune responses in UC.
We used SEB, a superantigen, to mimic the antigen-specific interactions between various immune cells. Treatment of SEB-stimulated PBMCs and B cells with IL-35 significantly increased the level of secreted IL-10 in UC patients. The two cytokines that shared subunits with IL-35, IL-12, and IL-27 did not portrait a similar role. Interestingly, we found that IL-35 suppressed the proliferation of T cells, including both CD4+CD25− conventional T cells and CD4+CD25+ Treg cells, as well as CD8+ T cells. Furthermore, presence or absence of CD4+CD25+ Treg cells did not significantly change the level of secreted IL-10 from B cells, suggesting that IL-35-mediated upregulation of B cell IL-10 did not require the presence or upregulation of Treg cell activity. IL-35 did not significantly suppress B cell proliferation. These studies together suggested that IL-35 suppressed T cell inflammation by inhibiting the proliferation of proinflammatory and anti-inflammatory T cells, while in B cells, IL-35 mediated IL-10 secretion, but did not act on proliferation.
Next, we observed that treating UC B cells with IL-35 significantly enhanced their IL-10-dependent regulatory function. IL-35-treated B cells suppressed the IFN-γ, IL-17, and TNF-α secretion and proliferation of autologous CD4+CD25− T cells and CD8+ T cells from UC patients, and effect that could be reduced by blocking IL-10 signaling. At the same time, we observed that CD4+CD25− T cells secreted high levels of IL-5 and IL-13, two cytokines upregulated in UC, after SEB stimulation. However, IL-35-treated B cells did not demonstrate an effect on regulating IL-5 and IL-13 secretion, which indicated that IL-35-treated B cells primarily mediated suppression of Th1 and Th17 inflammations.
These previous discoveries identified a potent regulatory role of IL-35. To investigate the in vivo activities of IL-35 in UC, we examined the IL-35 expression in IMCs from UC tissues. We found that IMCs from UC patients presented significantly lower secreted IL-35 than IMCs from healthy controls. Since the IL-35 subunits p35 and EBI3 were independently regulated, we examined the transcription of p35 and EBI3 (Jones et al., 2012). Interestingly, although EBI3 transcription level was not significantly different between UC and controls, the UC patients presented significantly lower p35 transcription. Since p35 expression is inhibited by Th2 transcription factor c-Maf (Homma et al., 2007), these results suggested that reduced IL-35 expression was due to a suppression of subunit p35 mediated by Th2 responses that were elevated in UC patients. IL-35 shares the p35 subunit with IL-12 and the EBI3 subunit with IL-27. It is yet unclear whether the other IL-12 and IL-27 subunits, namely IL-12 p40 and IL-27 p28, share the same expression trend as p35 and EBI3. It was previously shown that IL-27 p28 transcript level was significantly elevated in mucosa from Crohn's disease, but not UC patients (Schmidt et al., 2005). Further studies are required to examine the expression level of IL-12 p40 in the gut mucosa of UC patients and investigate the underlying regulatory pathways that control the expression of subunits in the IL-12 cytokine family.
The inflammatory and metabolic pathways are intertwined in the maintenance of homeostasis in intestinal mucosa. For example, the circulating level of glucagon-like peptide-2 (GLP-2), an endocrine peptide that is tropic to the intestinal epithelium, was dysregulated in UC (Xiao et al., 2000). Interestingly, treatment of rat IBD with GLP-2 had significantly reduced the severity of lesions and alleviated the level of inflammation, which was accompanied by significant decreases in the level of IFN-γ and TNF-α (Alavi et al., 2000). Similarly, we demonstrated the IL-35-treated B cells suppressed the IFN-γ, IL-17, and TNF-α responses of T cells. Therefore, in future studies, the therapeutic value of IL-35, as well as Breg cells in UC, should be examined.
Footnotes
Disclosure Statement
No competing financial interests exist.