
Abstract
Heat shock protein Hsp16.3 is closely related to latent Mycobacterium tuberculosis (MTB) infection and plays an important role in sustained survival when MTB is dormant. In this study, the Hsp16.3 gene mutant MTB H37Rv strain (Hsp16.3ΔMTB) was obtained through gene recombination and infected into murine RAW 264.7 macrophages. Western blotting and immunofluorescence showed increased expression of the autophagy-related protein LC3, and transmission electron microscopy showed significantly increased macrophage autophagosomes, suggesting that Hsp16.3ΔMTB facilitates murine macrophage autophagy. These findings have implications for preventing and controlling tuberculosis.
Introduction
D
Macrophages are the primary barrier against the spread of MTB in the body (Bao et al., 2016). However, it is peculiar that macrophages do not completely eliminate the phagocytosed MTB as they do other extracellular bacteria. Typically, MTB blocks phagosome maturation, preventing fusion with lysosomes. However, macrophages can initiate an autophagy pathway to promote phagosome maturation and lysosome fusion, resulting in the elimination of intracellular bacteria (Ní et al., 2011). Latent infection of MTB can adapt to the host immune response by regulating the expression of related genes to increase the synthesis of proteins that interfere with the autophagy pathway and prevent the autophagy-mediated transport of pathogens to lysosomes to prevent lysosomal degradation (Subbian et al., 2013), hence facilitating MTB replication and survival (Kim et al., 2012). Hsp16.3 expression is increased by 80-fold in latent infection compared with the level during active infection, conferring on MTB the ability to escape macrophage autophagy (Yuan et al., 1996). As an important latent infection, associated protein, Hsp16.3 might interfere with macrophage initiation of autophagy, preventing normal macrophage function, and thereby facilitate MTB survival in macrophages.
This study was mainly based on the principle of using homologous recombination to construct an Hsp16.3-mutated MTB (Hsp16.3ΔMTB) strain, which was then used to infect murine RAW 264.7 macrophages to study the effect of Hsp16.3 on macrophage autophagy, to provide a meaningful reference for preventing and controlling tuberculosis, especially in terms of the mechanism of latent infection.
Materials and Methods
Bacteria, plasmid, and cells
The MTB H37Rv strain was preserved at the Fourth Military Medical University Laboratory Animal Center (Zhang et al., 2012). The pKO plasmid was kindly provided by Professor Wanjiang Zhang (Shihezi University, Xinjiang, China). Mouse RAW 264.7 macrophages were purchased from ATCC (Rockville, MD).
Reagents
The restriction enzymes, Taq enzyme, and DNA marker were purchased from Takara (Dalian, China). The plasmid isolation, purification, and recovery kits were from Sangon Biotech (B518191, Shanghai, China). Kanamycin, bovine serum albumin, horseradish peroxidase (HRP)-goat anti-rabbit immunoglobulin G (IgG), and HRP-goat mouse IgG were from Shanghai Transhold Tech. Dev. (Shanghai, China). 7H9 broth, 7H10 agar, and oleic acid-albumin-dextrose-catalase (OADC) were from BD Biosciences (271310, 262710 and 211886, San Jose, CA). Rapamycin, 3-methyladenine (3-MA), and bafilomycin A1 (BAF) were from Sigma-Aldrich (V900930), Selleckchem (S2767, Houston, TX), and BBI Life Sciences (A601116, Amherst, NY), respectively. The antibodies against LC3 and Beclin 1 were from Marine Biological Laboratory (MBL) (PM036 and PD017, Woburn, MA). Atg5 antibody was purchased from CST (#2630, Danvers, MA).
Isolation of MTB genomic DNA
MTB H37Rv strain stored in Lowenstein-Jensen medium at 4°C was seeded in 7H9 medium with 10% acid-albumindextrose-catalase (ADC) and cultured in a 37°C incubator for 4 weeks. MTB genomic DNA was isolated according to the manufacturer's instructions (9763, TaKaRa Mini BEST bacteria genomic DNA extraction kit ver.3.0).
Primer synthesis and target gene amplification
Primers were designed according to the full-length sequence of the Hsp16.3 gene in GenBank and synthesized by Sangon Biotech. The primer sequences were as follows (underlined sections indicate restriction sites, followed by the restriction enzymes in parentheses): P1: 5′-TTAGGTACCCACCGTGGTCCGAGATT-3′ (KpnI); P2: 5′ATAGGATCCCCCG TGTACGTGCTGAAT-3′ (BamHI); P3: 5′-TTACTGCAGGGATAGCCGAGGACCACA-3′ (PstI); P4: 5′-TTA
Recombinant plasmid pKO-Hsp16.3 construction, identification, and sequencing
Target gene fragments (663 bp) obtained by KpnI and BamHI digestion were linked with the pKO vector, resulting in the positive clone pKO-Hsp16.3-5′. The 683-bp target gene fragments obtained by PstI and HindIII digestion were linked with pKO-Hsp16.3-5′ by T4 DNA ligase, resulting in the recombinant plasmid pKO-Hsp16.3. The recombinant plasmid was identified by digestion using two restriction enzymes and sequencing.
Preparation of MTB competent cells and electrotransformation
MTB H37Rv cells were inoculated into 7H9 medium containing 10% ADC. After 3 weeks of incubation at 37°C, a 0.1 M volume of 2 M glycine was added and the cells were incubated for 24 h, followed by incubation on ice for 1 h. The cells were harvested by centrifuging at 4000 rpm for 10 min. The supernatant was discarded and the cells were resuspended in ice-cold 10% glycerol, washed thrice by centrifuging at 4000 rpm for 10 min, resuspended with ice-cold 10% glycerol, and stored at −80°C. The competent cells were thawed on ice, mixed with 1 μg of recombinant plasmid pKO-Hsp16.3, transferred to a cuvette (0.4-cm gap; Bio-Rad), and electroporated (at 2.5 kV, 25 μF, 1000 Ω). Then, the cells were diluted with 10 mL of 7H9 medium containing 10% ADC and resuscitated by continuous shaking at 37°C for 16 h. Then, 200 μL of the cells were coated on a 7H10 agar plate containing 10% OADC and 30 μg/mL kanamycin and cultured at 37°C to form a single colony.
Hsp16.3 mutant strain screening and identification
A single colony was randomly selected from the 7H10 plates and cultured in 7H9 medium containing 10% ADC and 30 μg/mL kanamycin. After centrifugation, the pellet was boiled for 10 min, followed by centrifugation at 4000 rpm for 2 min. DNA (2 μL) was isolated from the cells as the template, and PCR was performed using primers P7 and P8; DNA isolated from normal MTB H37Rv was used as the control. The PCR conditions were as described above. The PCR products were separated by 1% agarose gel electrophoresis and cells with the 1232-bp fragment were marked. The marked strain was inoculated on a 7H10 plate containing 10% sucrose solution and 10% OADC and incubated at 37°C for 4 weeks. Single colonies were picked and inoculated in 7H9 medium containing 10% sucrose solution and 10% ADC, and then cultured by shaking for 4 weeks. The cells were harvested by centrifugation, and the pellet was boiled for 10 min, followed by centrifugation at 4000 rpm for 2 min. PCR was performed using primers P5 and P6, and normal MTB H37Rv was used as the control. The PCR conditions were as described above. The colony without the 435-bp PCR product was the mutant strain.
Effects of rapamycin and 3-MA on RAW 264.7 cell autophagy
RAW 264.7 cells were treated with 0, 10, 50, or 100 nmol/L rapamycin for 6 h and analyzed by Western blotting to determine the changes in LC3 expression. Briefly, aliquots of cell lysates containing protein were separated by 12% sodium dodecyl sulfate–polyacrylamide gel electrophoresis and transferred to nitrocellulose filters. The filters were blocked with TBST buffer containing 5% skimmed milk and incubated with anti-LC3 antibody or anti-Beclin1 antibody overnight, followed by the addition of HRP-linked anti-mouse IgG and electrochemiluminescence visualization of the bands. Based on the results obtained, RAW 264.7 cells were treated with 100 nmol/L rapamycin for 6 h, followed by 24-h treatment with 10, 25, 40, or 60 μmol/L 3-MA. LC3 and Beclin1 levels were determined by Western blotting as described above.
Effect of mutant Hsp16.3 gene on RAW 264.7 cell autophagy
RAW264.7 macrophages were cultured in RPMI-1640 medium (1786004; GIBCO, Thermo Scientific) supplemented with 10% fetal bovine serum (FBS; SV80087.01, HyClone, Thermo Scientific) and 1% penicillin/streptomycin (P1400; Solarbio, Beijing). For infection, RAW 264.7 cells (1 × 105) were cultured in 6-well plates for 24 h and divided into five groups: those infected with normal MTB H37Rv or Hsp16.3ΔMTB for 24 h (infection groups); those infected with Hsp16.3ΔMTB followed by 6-h treatment with 100 nmol/L rapamycin (RAPA group); those infected with Hsp16.3ΔMTB followed by 6-h treatment with 100 nmol/L rapamycin and 24-h treatment with 60 μmol/L 3-MA (3-MA group); and those infected with Hsp16.3ΔMTB followed by 4-h treatment with 500 nmol/L BAF (BAF group). The bacteria were resuspended in RPMI 1640 medium and infected at 104 colony-forming units (CFU). The protein samples were prepared according to the manufacturer's instructions.
Confocal microscopy
RAW 264.7 cells (1 × 105) were seeded on a plate for confocal microscopy. After 24 h of incubation, the macrophages were infected with 104 CFU Hsp16.3ΔMTB for 24 h and treated with rapamycin or 3-MA or BAF, according to the above mentioned method. Then, the plate was washed twice with phosphate-buffered saline (PBS). Treated cells were fixed in 4% paraformaldehyde for 30 min, permeabilized with 0.1% Triton X-100, and then incubated with anti-LC3 antibody overnight. The cells were washed twice with PBS and then incubated with 200 μL of fluorescent secondary antibody (1:500) for 30 min. Next, the cells were washed twice with PBS buffer again and the intensity of LC3 puncta was quantified by confocal microscopy.
Transmission electron microscopy
Autophagosome formation was observed using transmission electron microscopy. Macrophages in each group were washed thrice with PBS, prefixed with 3% glutaraldehyde, and fixed with 10 g/L osmium tetroxide overnight. Ultrathin sections were stained with uranyl acetate and lead citrate and observed under a Hitachi H-7100 transmission electron microscope (Hitachi High-Technologies, Tokyo, Japan). For each group, 100 cells were observed to determine the autophagosome ratio.
Knockdown of atg5 expression in RAW 264.7 macrophages
RAW 264.7 macrophages were plated in a 24-well plate at a density of 0.5 × 105 cells per well, 24 h before viral infection. Mouse atg5 siRNA Lentivirus (ABM, Inc., Richmond, BC, Canada) was transiently transfected into cells in the wells according to the manufacturer's instructions. A complete optimal medium (0.5 mL with 10% FBS and polybrene at a concentration of 8 μg/mL) was added and cells were incubated at 37°C with 5% CO2 overnight. The culture medium was removed and replaced with 1 mL of complete medium. The cells were then incubated at 37°C with 5% CO2 overnight. The downstream expression of atg5 was assayed by Western blot analysis.
Determination of intracellular bacterial viability
The multiplicity of infection of the Hsp16.3ΔMTB-infected macrophages was 1:10. Then, the bacteria were incubated at 37°C for 24 h. The suspension was collected for inoculation in 7H10 agar containing 100 g/L OADC and incubated at 37°C. After 4 weeks, the CFUs per mL suspension were counted, and the bacterial activity in the macrophages was set as log10 CFU. All experimental groups were seeded in six parallel wells.
Statistical analysis
Data are the mean ± standard deviation of three independent experiments performed in triplicate. The least significant difference t-test was used for statistical analysis of the differences in the experimental results; p < 0.05 indicated statistical significance.
Results
Double enzyme digestion and sequencing of recombinant plasmid pKO-Hsp16.3
Two DNA fragments (663 and 684 bp) from either end of the MTB Hsp16.3 gene were obtained by double enzyme digestion of the recombinant plasmid pKO-Hsp16.3, and sequencing showed that they were of the same size as the 5′ and 3′ terminal sequences of the Hsp16.3 gene from the MTB H37Rv strain (Fig. 1A, B). The sequencing results are shown in the Supplementary Figures S1–S3 (Supplementary Data are available online at

PCR screening to identify Hsp16.3ΔMTB-positive cells.
PCR screening of Hsp16.3ΔMTB
Positive clones screened from the kanamycin-containing 7H10 agar plates were cultured in 7H9 broth containing 10% sucrose, and the bacterial DNA was amplified using primers P7 and P8. The strain with the 1232-bp fragment (Kana gene) was noted as the positive strain, and the fragment was further amplified using primers P5 and P6. Colonies without the 435-bp fragments (Hsp16.3) were the Hsp16.3-mutated MTB strain. We designated colony 2 of these strains as Hsp16.3ΔMTB (Fig. 1C, D) and used it in the subsequent analysis.
Effects of rapamycin and 3-MA on autophagy activity in macrophages
When mouse RAW 264.7 cells were treated with 100 nmol/L rapamycin, the expression of the autophagy-related protein LC3II increased significantly by Western blot analysis (Fig. 2A, B). However, when the macrophages were treated with 100 nmol/L rapamycin for 6 h and then with 60 μmol/L 3-MA for another 24 h, LC3II expression level was significantly decreased. This finding indicated that the autophagy induced by rapamycin in RAW 264.7 macrophages was markedly inhibited by 3-MA (Fig. 2C, D).
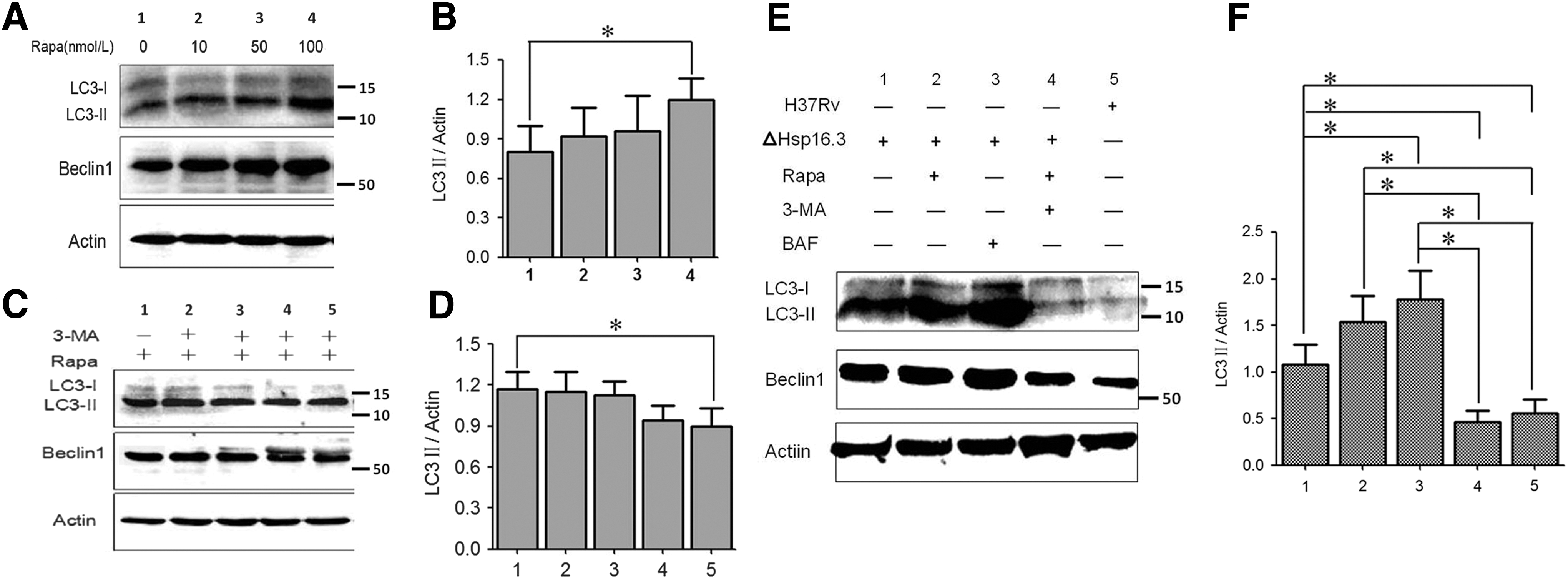
LC3 and Beclin1 expression in RAW 264.7 cells induced by Hsp16.3ΔMTB infection.
Effect of Hsp16.3ΔMTB on LC3 and Beclin1 expression levels
An increased LC3II expression was induced in RAW 264.7 cells by Hsp16.3ΔMTB infection, and the expression level was significantly higher than that in the MTB H37Rv infection group. The addition of 100 nmol/L rapamycin further increased the level of autophagy, but the addition of 60 μmol/L 3-MA significantly decreased LC3II expression (Fig. 2E, F). The expression of Beclin1 displayed a similar trend. When RAW 264.7 cells were infected with Hsp16.3ΔMTB in the presence of BAF, autophagy degradation was effectively inhibited, and the level of LC3II/actin was higher than that in cells infected with the Hsp16.3-knockout mutant without BAF treatment (Fig. 2E, F).
Effect of Hsp16.3ΔMTB on LC3 puncta in RAW 264.7 cells
Hsp16.3ΔMTB infection of RAW 264.7 cells caused numerous LC3 puncta in the cytoplasm (Fig. 3A), suggesting that more autophagic flux had occurred. Both the number and intensity of LC3 puncta peaked after the addition of rapamycin, and were significantly higher than those of RAW 264.7 cells infected with MTB H37Rv (Fig. 3B). However, the autophagy inhibitor 3-MA significantly decreased the number and intensity of the intracellular LC3 puncta (Fig. 3C) and BAF inhibited autophagic degradation, which resulted in abundant LC3 puncta aggregates (Fig. 3E, F).

Effects of Hsp16.3ΔMTB on LC3 puncta in RAW 264.7 cells.
Autophagosome formation
When RAW 264.7 cells were infected with Hsp16.3ΔMTB, a large number of autophagosomes in the cytoplasm were observed by transmission electron microscopy (Fig. 4A), and the addition of rapamycin further facilitated the formation of autophagosomes in the macrophages (Fig. 4B). However, the addition of 3-MA significantly reduced autophagosome formation (Fig. 4C). Furthermore, the typical MTB phagocytic structure (mycobacterial phagosomal compartments) was formed in macrophages infected with MTB H37Rv (Fig. 4D). In the BAF treatment group, abundant accumulation of autophagosomes in the organelles and cytoplasm components was found, whereas very few autolysosomes were observed (Fig. 4E, F).
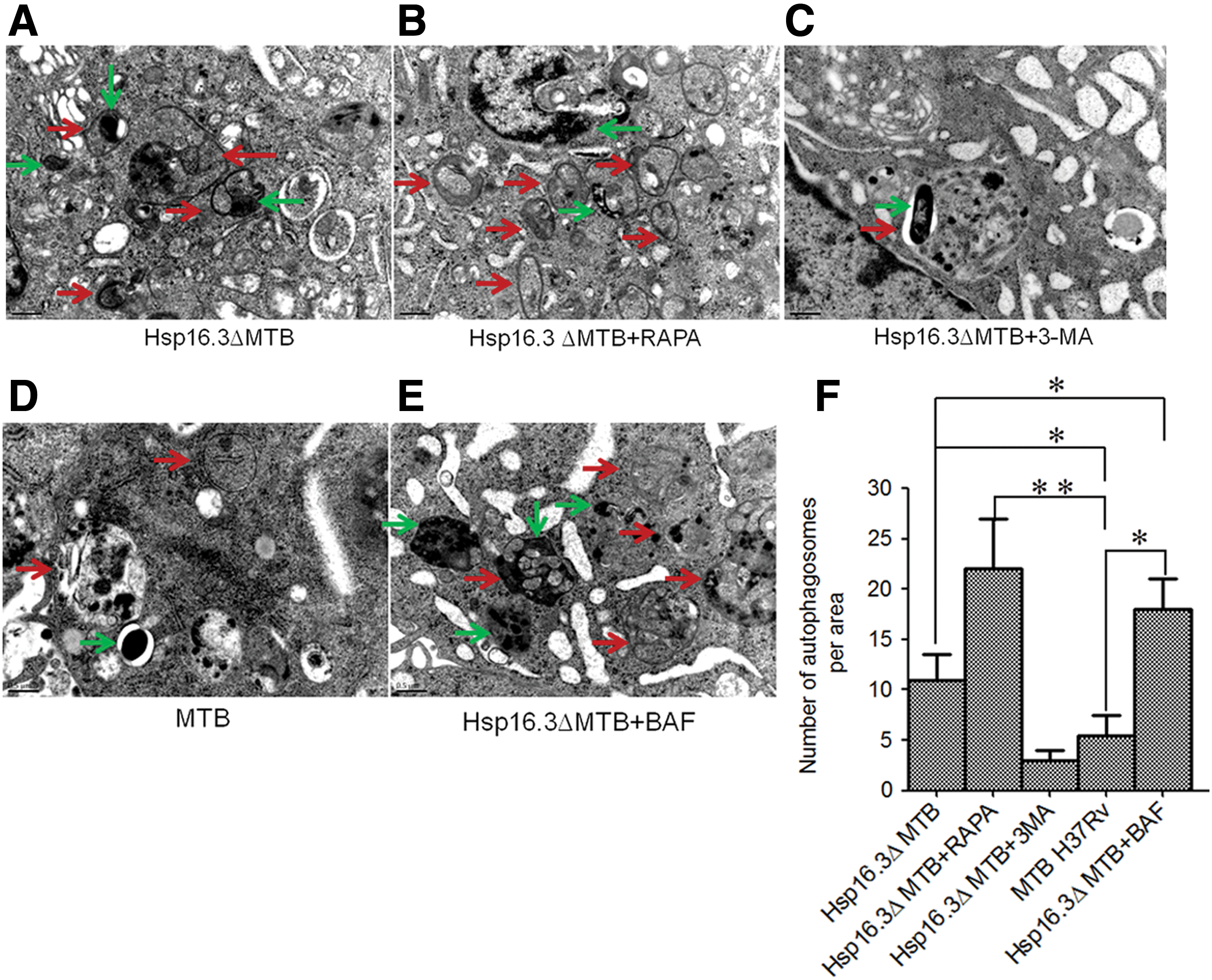
Effects of Hsp16.3ΔMTB on autophagosome formation in RAW 264.7 cells.
MTB infection status of macrophages
Infected RAW 264.7 cells were detected by acid-fast staining. The morphology of bacteria phagocytosed by macrophages is shown in Supplementary Figure S4. Most bacteria were accumulated in macrophages infected with MTB H37Rv, whereas conspicuously fewer bacteria were presented in the Hsp16.3ΔMTB group. The number of bacteria further decreased with the addition of rapamycin, but the addition of 3-MA was followed by the appearance of a large number of MTB.
CFUs of intracellular bacteria
MTB-infected RAW 264.7 cells were digested and then incubated in 7H10 culture medium at 37°C for 28 days to count the CFUs (Fig. 5A). The initial log10 CFU of phagocytosed bacteria (Hsp16.3ΔMTB) at 0 h reached 7.88 ± 0.43. However, the log10 CFU of macrophages infected with MTB H37Rv after incubation for 24 h was 5.40 ± 0.51, whereas that of the Hsp16.3ΔMTB group was 4.02 ± 0.40 (p < 0.05), indicating that knockdown of Hsp16.3 expression promoted MTB elimination from the cells by autophagy. The log10 CFUs of the 3-MA group and rapamycin group were 6.50 ± 0.31 and 3.20 ± 0.32, respectively (Fig. 5A).
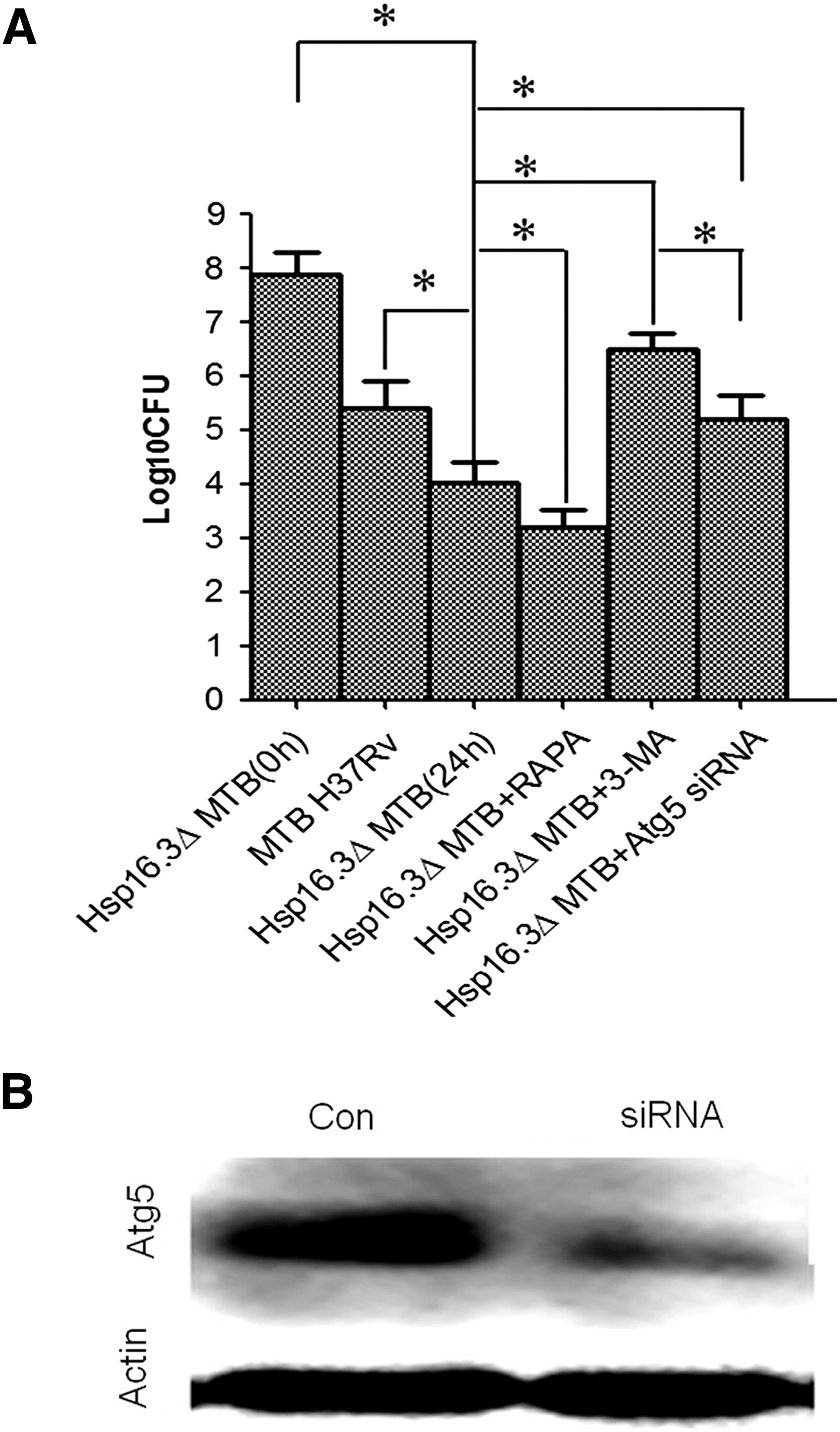
CFUs of Hsp16.3ΔMTB phagocytosed by RAW 264.7 cells.
To assess bacterial survival in cells lacking the autophagy pathway, we treated RAW 264.7 cells with mouse atg5-targeting siRNA. Western blot analysis showed that the expression of atg5 was effectively decreased (Fig. 5B). After infection with Hsp16.3ΔMTB, the CFUs in macrophages treated with atg5-targeting siRNA were counted. CFU value was decreased significantly in the Hsp16.3ΔMTB infection group compared with the MTB H37Rv infection group. In order to support the claim that this decrease was induced by autophagy, we knockdown the autophagy relative molecule atg5 expression in macrophages and infected with Hsp16.3ΔMTB. The results showed the CFUs value increased, meaning this decrease was reversed (Fig. 5A). This demonstrated that knockdown of atg5 expression in macrophages could induce CFUs number increase significantly in the Hsp16.3ΔMTB infection group (p < 0.05), suggesting that bacterial survival of Hsp16.3-mutated MTB could be rescued in the treated macrophages.
Discussion
Hsp16.3 is an alpha-crystallin-type Hsp, consisting of 144 amino acids with a molecular weight of ∼16.3KD (Shi et al., 2009). It is a key gene for the replication of MTB in macrophages (Meng et al., 2014). Early research has demonstrated that recombinant MTB H37Rv with Hsp16.3 gene deletion could only grow very small clones when cultured on 7H10 solid agar plates for ∼8–10 weeks. However, these clones could survive for 10 weeks (Stein, 2011), which indicated that Hsp16.3 plays a role in regulating sustained MTB growth (Yuan et al., 1998). Gutierrez et al. (2005) and Amano et al. (2006) found that autophagy is closely related to the survival of MTB in macrophages and macrophage autophagy could effectively eliminate the intracellular retention of MTB (Min et al., 2010). All of these results indicate that Hsp16.3 protein secretion and the macrophage autophagy pathway are closely related in MTB-infected macrophages, which may have a regulatory effect on the metabolism of dormant MTB.
The pKO vector can replicate in Escherichia coli, but it cannot be amplified in Mycobacterium owing to the lack of a replication origin, rendering it a good carrier for gene knockout (Arthur et al., 1987). In this study, two DNA fragments (663 and 683 bp) on either side of MTB Hsp16.3 were amplified and inserted into the corresponding sites in the pKO vector, followed by electroporation to incorporate the recombinant plasmid into MTB competent cells. After repeated screening with kanamycin sulfate and sucrose, an MTB strain with the Kana gene and without the Hsp16.3 gene fragment was obtained and inoculated into sucrose-containing Sutong liquid medium to obtain a mutant strain of single clonal origin.
Autophagy is regulated by autophagy-related proteins, including Atg5, Atg7, Atg8, and Atg12, which are involved in autophagosome formation (Pilli et al., 2012; Woo et al., 2014; Murrow et al., 2015; Vij et al., 2016). Among them, Atg8 plays an important role in regulating autophagy (Biswas et al., 2008; Lévêque et al., 2016); LC3 in mammals is an important marker of autophagy, and its expression can be used to detect autophagosome formation (Chandra and Kumar, 2016; Dong et al., 2016). In this study, LC3II expression levels increased markedly following rapamycin treatment. Western blotting showed significantly increased LC3II expression levels in macrophages following infection with Hsp16.3ΔMTB. Confocal microscopy of the infected macrophages showed increased LC3 fluorescent puncta, and numerous autophagy capsules were observed by transmission electron microscopy. This clearly demonstrated the formation of autophagosomes, which was significantly reduced by 3-MA treatment. When RAW 264.7 cells were infected with Hsp16.3ΔMTB in the presence of BAF, the levels of LC3II/actin and LC3II puncta were higher than those in cells infected with Hsp16.3ΔMTB without BAF treatment, suggesting that infection with the Hsp16.3-knockout mutant induces autophagy in host cells.
To support the claim that Hsp16.3 deletion promoted MTB elimination from the cells by autophagy, atg5-knockdown RAW 264.7 cells were infected with Hsp16.3ΔMTB to improve bacterial survival and increase bacterial CFUs. We hypothesize that in MTB-infected mouse macrophages, Hsp16.3 interferes with LC3 expression to disrupt autophagosome formation, thereby inhibiting MTB elimination by macrophages, which may be an important mechanism in the immune escape of latent MTB infection.
In summary, infection of RAW 264.7 macrophages with the mutant Hsp16.3 MTB strain H37Rv increased the expression of the autophagy-related protein LC3 and significantly promoted the formation of macrophage autophagosomes, which facilitate macrophage autophagy. At present, the Hsp16.3 pathway and interaction of Hsp16.3 with cytokines to regulate autophagy have not been clearly elucidated; therefore, further investigations warranted in this regard. These findings have implications for preventing and controlling tuberculosis, especially by clarifying the mechanism of latent infection.
Footnotes
Acknowledgments
This work was supported by the National Natural Science Foundation of China Program No. 31501112 and Shaanxi Natural Science Foundation Program No. 2016FWPT-02.
Disclosure Statement
No competing financial interests exist.
References
Supplementary Material
Please find the following supplemental material available below.
For Open Access articles published under a Creative Commons License, all supplemental material carries the same license as the article it is associated with.
For non-Open Access articles published, all supplemental material carries a non-exclusive license, and permission requests for re-use of supplemental material or any part of supplemental material shall be sent directly to the copyright owner as specified in the copyright notice associated with the article.