
Abstract
Small nucleolar RNA host gene 12 (SNHG12) is a novel long noncoding RNA identified to be upregulated and functions as an oncogene in several cancers. However, the function of SNHG12 and its target genes in modulating nonsmall cell lung cancer (NSCLC) development are rarely reported. In the present study, we validated that SNHG12 was overexpressed, while miR-138 was low-expressed, in NSCLC cells compared with normal human lung epithelial cells. SNHG12 harbored the binding site of miR-138 and inversely regulated the expression miR-138. Knockdown of SNHG12 inhibited proliferation and colony-forming ability, induced apoptosis, and increased caspase-3 activity of NSCLC cells, whereas miR-138 downregulation restored these effects. Furthermore, SNHG12 knockdown decreased volumes and weight of xenograft tumors in a NSCLC mouse model. Taken together, these findings suggested that knockdown of SNHG12 suppressed cell growth and induced apoptosis by upregulating miR-138 in NSCLC.
Introduction
L
miRNAs are groups of small, noncoding RNAs (19–23 nucleotides) that negatively regulate the expression of protein-coding genes by binding 3′-untranslated region (3′-UTR) of targeted genes (Tian et al., 2016). Accumulating evidence indicates that dysregulated miRs have been extensively studied, and their functions in diverse biological processes, such as proliferation, differentiation, metastasis, and apoptosis, which are crucial in the development of cancer, have been elucidated in multiple cancers, including NSCLC (Lin and Gregory, 2015; Han et al., 2016). Among the NSCLC-associated miRNAs, miR-138, a well studied tumor suppressor, was previously shown to be frequently downregulated in NSCLC cells and tissues (Ye et al., 2015; Xiao et al., 2016). We adopted an online bioinformatics database (Starbase v2.0) to predict potential SNHG12 target miRNAs. Among them, miR-138 was found to contain conserved target site of SNHG12. Thus, we hypothesized that SNHG12 exerted its oncogenic activity by sponging miR-138. The purpose of this study was to test this ceRNA hypothesis in NSCLC.
Materials and Methods
Cell lines and cell transfection
Five human lung cancer cell lines (H1299, A549, H358, H1975, and SPC-A1) and a normal human lung bronchial epithelial cell line (16-HBE) were purchased from the Chinese Academy of Sciences Cell Bank (Shanghai, China). All cells were cultured in RPMI-1640 medium (Gibco, Carlsbad, CA) supplemented with 10% fetal bovine serum (Gibco), 100 U/mL penicillin, and 100 μg/mL streptomycin in a humid atmosphere containing 5% CO2 at 37°C. All cell lines were verified as mycoplasma free.
miR-138 mimics (miR-138), scrambled miRNA oligonucleotides (miR-NC), miR-138 inhibitor (anti-miR-138), and inhibitor scrambled negative control (anti-miR-NC) were purchased from GenePharma (Shanghai, China). The nucleotide sequences of siRNA for SNHG12 were as follows: sense 5′-GCA GUG UGC UAC UGA ACU UTT-3′ and antisense 5′-AAG UUC AGU AGC ACA CUG CTT-3′. si-SNHG12 and nonspecific negative control siRNA (si-NC) were obtained from GenePharma. Cells were cultured and transfected with miRNAs, si-SNHG12, or si-NC using Lipofectamine 2000 (Invitrogen, Carlsbad, CA) following the manufacturer's instructions. The shRNA targeting SNHG12 (sh-SNHG12) and a negative control (sh-NC) were synthesized and cloned into the pENTR-U6 vector (Invitrogen).
Quantitative real-time PCR
Total RNA samples were extracted from cultured cells using the TRIzol reagent (Invitrogen) according to the manufacturer's instructions. cDNAs were synthesized from 1 μg of total RNA by using Prime-Script™ One Step RT-PCR Kit (TaKaRa, Dalian, China). SNHG12 was determined using a SYBR Premix Ex Taq II Kit (TaKaRa) on an Applied Biosystems 7500 Real-Time PCR system (Applied Biosystems, Foster City, CA). Expression of mature miR-138 was determined by the TaqMan miRNA-assay (Applied Biosystems). The expression levels of SNHG12 were normalized to GAPDH, and the miR-138 expression levels were normalized to U6. SNHG12 primers were forward: 5′-TCT GGT GAT CGA GGA CTT CC-3′ and reverse: 5′-ACC TCC TCA GTA TCA CAC ACT-3′. U6 primers were forward: 5′-GCT TCG GCA GCA CAT ATA CTA AAA T-3′ and reverse: 5′-CGC TTC ACG AAT TTG CGT GTC AT-3′; GAPDH primers were forward: 5′-CGC TCT CTG CTC CTC CTG TTC-3′ and reverse: 5′-ATC CGT TGA CTC CGA CCT TCA C-3′.
Cell proliferation assay
The Cell Counting Kit-8 (CCK-8) assay was performed according to the manufacturer's protocols. In brief, after transfection, cells were seeded in 96-well plates and incubated for 24, 48, and 72 h in a humidified incubator at 37°C and 5% CO2. Specifically, 10 μL of CCK-8 assay solution was added to each well and further incubated for 2 h. The optical density of each well was read by a microplate reader (Molecular Devices, Sunnyvale, CA) at 450 nm to estimate cell proliferation.
Colony formation assay
A certain number of transfected cells were seeded into six-well plates and incubated for 10 days. The colonies were fixed with 4% formaldehyde (Bogoo, Shanghai, China) for 15 min and stained with 0.1% crystal violet (Solarbio, Beijing, China) for 5 min. The number of colonies counted to evaluate the colony-forming ability.
Flow cytometry analysis
Transfected cells were harvested after transfection for 48 h. After the double staining with 2.5 μL Annexin V-FITC and 5 μL propidium iodide at room temperature for 10 min using the FITC Annexin V Apoptosis Detection Kit (BD Biosciences, San Jose, CA), the apoptotic cells were analyzed by FACScan Flow Cytometry (BD Biosciences).
Caspase-3 activity assay
Activity of caspase-3 was detected using the Caspase-3 Activity Kit (Beyotime Biotechnology, Haimen, China) according to the manufacturer's instructions. The absorbance was measured on a microplate reader (Molecular Devices) at 405 nm. Caspase-3 activity was measured after 48 h of transfection.
Luciferase reporter assay
Human H1299 and A549 cells (2.0 × 104) were cultured in 96-well plates and cotransfected with 100 ng of pmir-GLO-SNHG12-WT (SNHG12-WT) or pmir-GLO-SNHG12-MUT (SNHG12-MUT) (Sangon Biotech, Shanghai, China) and 20 ng of Renilla luciferase control vector (pRL-TK; Promega, Madison, WI) with 50 nM miR-183 mimic, anti-miR-183, or their scrambled negative control into cells using Lipofectamine 2000 (Invitrogen). The relative luciferase activity was measured using the dual-luciferase reporter assay system (Promega) and normalized to Renilla luciferase activity after incubating for 48 h.
RNA immunoprecipitation assay
RNA immunoprecipitation (RIP) assay was conducted to examine whether SNHG12 interacts with miR-138 in lung cancer cells using Magna RIP RNA-Binding Protein Immunoprecipitation Kit (Millipore, Billerica, MA) following the manufacturer's protocol. H1299 cells at 80–90% confluency were lysed in RIP lysis buffer, after which 100 μL of whole-cell extract was incubated with RIP buffer containing magnetic beads conjugated with human anti-Argonaute2 (Ago2) antibody (Millipore), normal mouse immunoglobulin G (IgG; Millipore) as a negative control, and anti-snRNP70 (Millipore) as a positive control for the RIP procedure. The protein in the precipitate was incubated with proteinase K buffer (Sigma-Aldrich), and the immunoprecipitated RNA was isolated. Purified RNA was analyzed by quantitative real-time PCR (qRT-PCR) to confirm the presence of the binding targets.
In vivo tumor formation assay
Animal experiments were performed with the approval of Institutional Animal Care and Use Committee of Huaihe Hospital of Henan University and in conformity with national guidelines for the welfare of laboratory animals. Four-week-old male BALB/c athymic nude mice were purchased from SLAC Laboratory Animal Center (Shanghai, China). Mice were housed and maintained in a specific pathogen-free facility and fed ad libitum. For the in vivo study, 5 × 106 H1299 cells transfected with sh-SNHG12 or sh-NC were subcutaneously injected into the right flank of the nude mice (n = 6 mice/group). Tumor volumes were monitored (0.5 × length × width2) every 7 days. After 5 weeks, the nude mice were sacrificed and tumors were weighted. The resected tumor tissues were snap frozen in liquid nitrogen and stored at −80°C for total RNA extraction.
Statistical analysis
Statistical analysis was performed using GraphPad Prism 5.0 (GraphPad Prism, San Diego, CA). The differences were analyzed by Student's t-test or one-way ANOVA. p < 0.05 was defined as statistically significant.
Results
Direct binding between SNHG12 and miR-138
To address the molecular mechanism by which SNHG12 exerts antitumor effects, bioinformatic analyses were carried out using the online bioinformatics databases (Starbase v2.0) aiming to screen the possible miRNAs that direct bind to SNHG12. Among the prediction results, miR-138 captured our attention since miR-138, a tumor suppressor, has been reported to be downregulated in NSCLC (Ye et al., 2015; Xiao et al., 2016), contrary to the expression status of SNHG12 in NSCLC. Figure 1A showed that the SNHG12 RNA contained one target site of miR-138. To validate early observation from previous reports that SNHG12 was upregulated (Iyer et al., 2015; Wang et al., 2017b) and miR-138 was downregulated in NSCLC (Ye et al., 2015; Xiao et al., 2016), the expression levels of SNHG12 and miR-138 in human NSCLC cell lines were analyzed using qRT-PCR. As shown in Figure 1B, SNHG12 expression was increased in five NSCLC cell lines (H1299, A549, H358, SPC-A1, and H1975) compared with that in normal human bronchial epithelial cell line 16-HBE (control). miR-138 expression was downregulated in NSCLC cell lines relative to that in 16-HBE (Fig. 2C). The status of SNHG12 and miR-138 expression was representative in H1299 and A549, thus, the two cell lines were selected in the following studies. To verify the interaction between SNHG12 and miR-138, dual-luciferase reporter assay was performed. The results revealed that overexpression of miR-138 significantly reduced the luciferase activities from the reporter SNHG12-WT, but not mutant reporter SNHG12-MUT in H1299 cells (Fig. 1D). Contrarily, downregulation of miR-138 increased the luciferase activities in A549 cells cotransfected anti-miR-138 and SNHG12-WT rather than those in cells transfected with anti-miR-Con and SNHG12 without miR-138 target site (SNHG12-MUT) (Fig. 1E). It is well known that miRNAs exert their gene silencing functions through forming RNA-induced silencing complex (RISC), of which core component was Ago2 (Tian et al., 2017). To determine whether SNHG12 and miR-138 are in the same RISC, RIP assay was conducted in H1299 cells using specific antibody against Ago2 protein. The results showed that SNHG12 and miR-138 were significantly enriched by about 524- and 669-fold in Ago2-containing immunoprecipitates relative to control IgG immunoprecipitates (Fig. 1F), consistent with the dual-luciferase reporter assay. To determine whether SNHG12 decreased the miR-138 levels, H1299 and A549 cells were transfected with si-NC or si-SNHG12, and the levels of miR-138 were detected by qRT-PCR. As shown in Figure 1G and H, the SNHG12 expression was significantly downregulated after transfection with si-SNHG12. The levels of miR-138 were increased in H1299 and A549 cells transfected with si-SNHG12 (Fig. 1I, J). Taken together, our results revealed that SNHG12 acted as an endogenous sponge “antagomir,” which can inversely regulate the expression of miR-138.
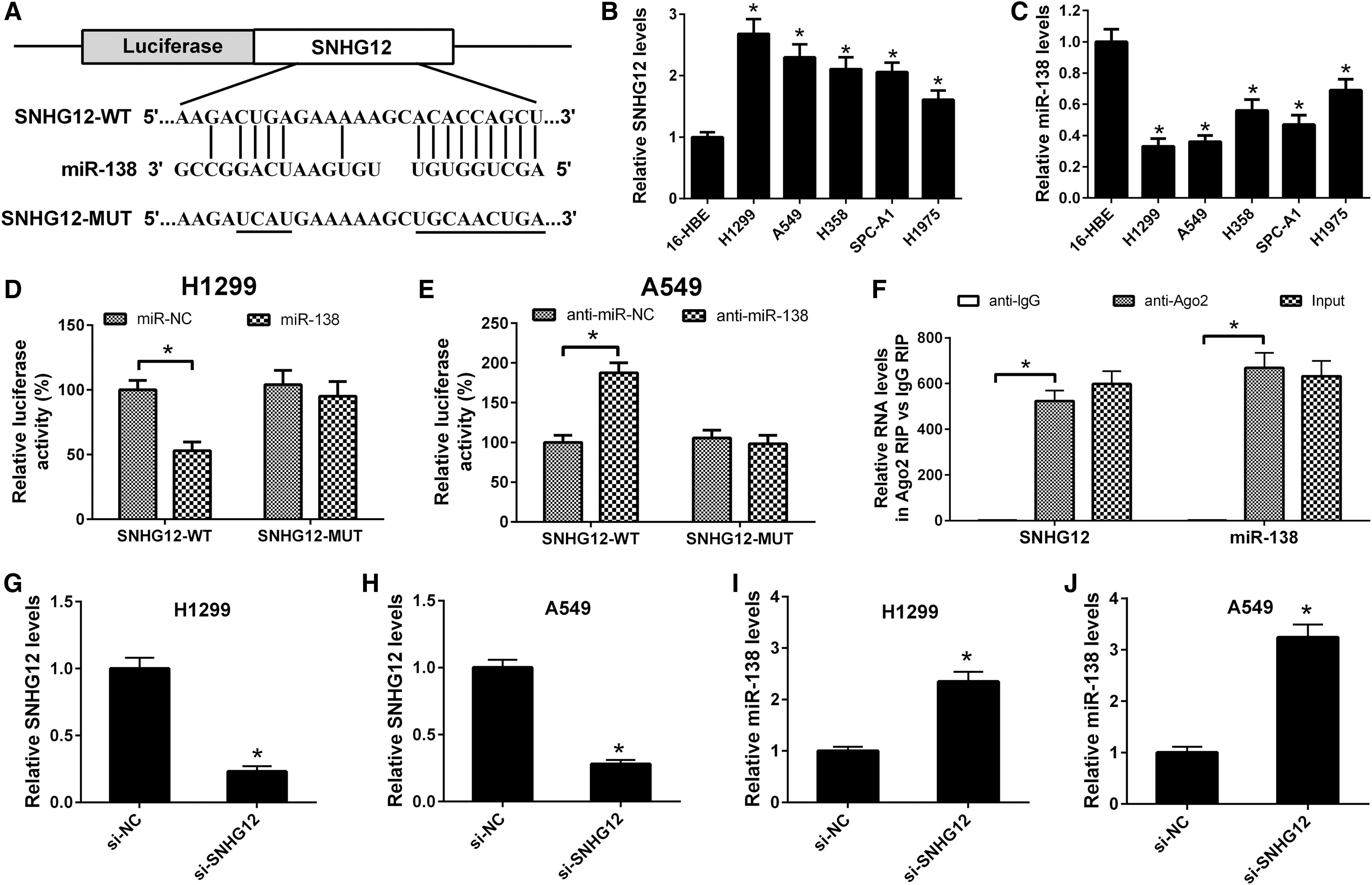
miR-138 was identified as a target of SNHG12.

SNHG12 knockdown inhibited NSCLC cell growth via upregulated miR-138.
Knockdown of SNHG12 inhibited NSCLC cell growth via targeting miR-138
To investigate whether SNHG12 knockdown inhibited NSCLC cell growth via targeting miR-138, we first confirmed the anticancer effects of miR-138 in NSCLC cells. As shown in Figure 2A and B, overexpression of miR-138 suppressed the proliferation of H1299 and A549 cells. Then cells were transfected with si-SNHG12 or cotransfected with si-SNHG12 and anti-miR-138, and the cell viability was assessed by CCK-8 assay after 24, 48, and 72 h of transfection. SNHG12 knockdown significantly suppressed the proliferation of H1299 and A549 cells, but miR-138 downregulation partially or completely restored this inhibitory effect (Fig. 2C, D). The effect of miR-138 overexpression on colony-forming ability of NSCLC cells was also evaluated using colony formation assay. As shown in Figure 2E and F, overexpression of miR-138 significantly reduced the number of colonies of H1299 and A549 cells. The colony number in the si-SNHG12 group was decreased compared with that in the si-Con group. Interestingly, miR-138 downregulation completely restored the suppression of colony-forming ability caused by SNHG12 knockdown in H1299 and A549 cells (Fig. 2G, H). These findings suggested that knockdown of SNHG12 inhibited NSCLC cell growth through targeting miR-138.
Knockdown of SNHG12 induced apoptosis of NSCLC cells by targeting miR-138
The role of miR-138 on apoptosis in NSCLC cells was determined using flow cytometry analysis. As shown in Figure 3A and B, miR-138 overexpression induced apoptosis of H1299 and A549 cells when compared with the miR-Con group. Moreover, caspase-3 activity was increased in the miR-138 overexpression group compared with the miR-Con group (Fig. 3C, D). To explore whether SNHG12 knockdown induced apoptosis of NSCLC cells by upregulating with miR-138, a restoration experiment was carried out in H1299 and A549 cells. Flow cytometry analysis showed that SNHG12 knockdown significantly increased apoptosis rates of H1299 and A549, but this was reversed by cotransfection of si-SNHG12 and a miR-138 inhibitor (anti-miR-138) in H1299 and A549 cells (Fig. 3E, F). Furthermore, caspase-3 activity assay suggested that caspase-3 activity was significantly increased in the SNHG12 knockdown group compared with the control group, but this was reversed by cotransfection of si-SNHG12 and anti-miR-138 in H1299 and A549 cells (Fig. 3G, H). Taken together, these results indicated that knockdown of SNHG12 induced apoptosis of NSCLC cells by upregulating with miR-138.
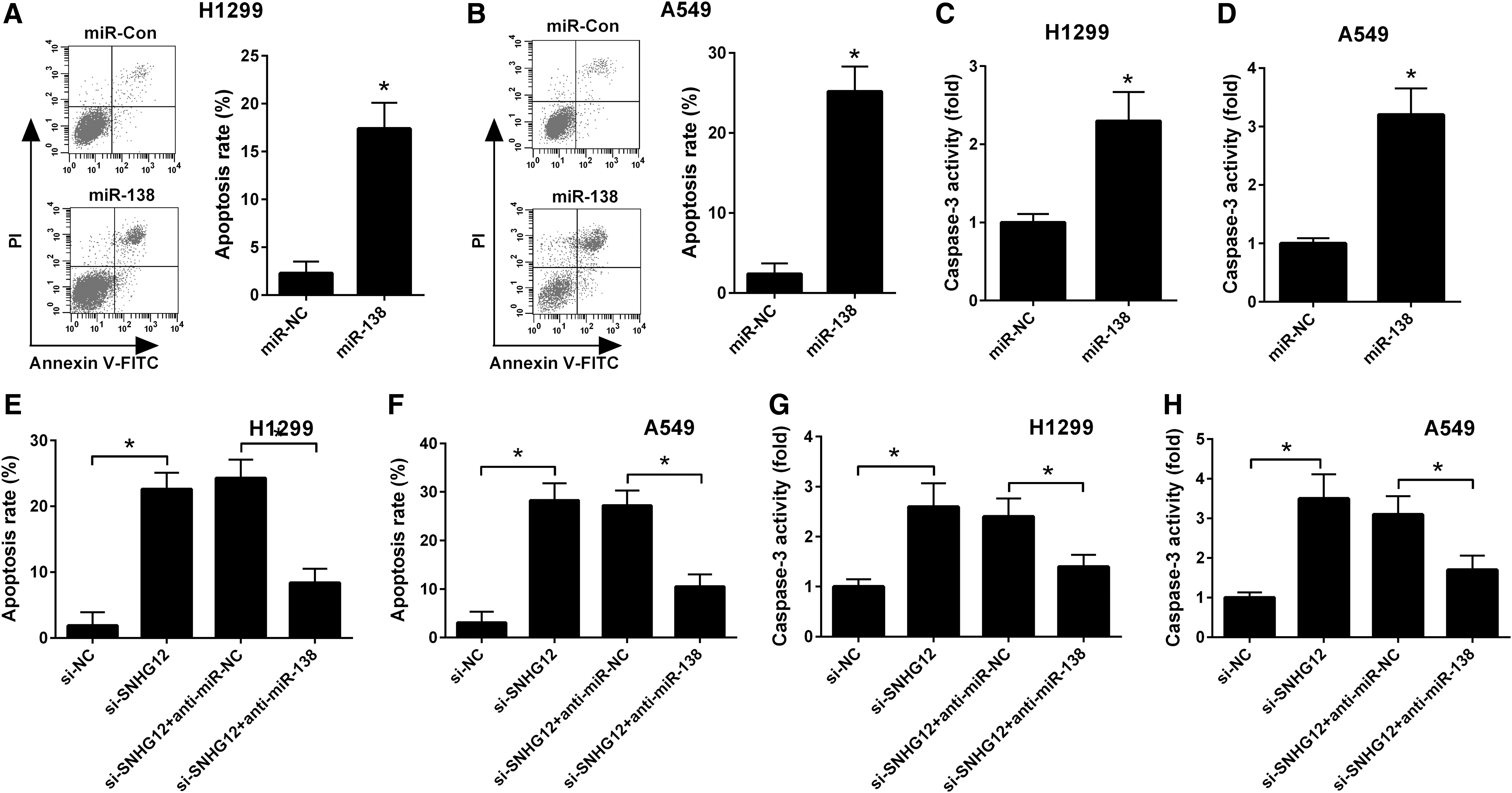
SNHG12 knockdown induced NSCLC cell apoptosis via upregulated miR-138.
SNHG12 knockdown inhibited xenograft tumor growth in a NSCLC mouse model
To further confirm the function of SNHG12 on tumorigenicity in vivo, H1299 cells transfected with sh-SNHG12 or sh-NC were subcutaneously injected into the right flank of the nude mice, and tumor volumes were monitored every 7 days. The tumors were collected and weighed after 5 weeks. SNHG12 knockdown significantly caused a decrease in tumor volume (Fig. 4A) and weight (Fig. 4B) when compared with the sh-NC group. To reveal the molecular mechanisms of SNHG12 in tumorigenicity, total RNAs were extracted from the resected tumor tissues to perform qRT-PCR analysis. The expression levels of SNHG12 were decreased in tumor tissues in the sh-SNHG12 group (Fig. 4C), whereas, the expression of miR-138 was upregulated in xenografts of the sh-SNHG12 group when compared with the sh-NC group (Fig. 4D). These results suggested that SNHG12 knockdown suppressed NSCLC tumor growth in vivo partially via upregulating tumor suppressor miR-138.
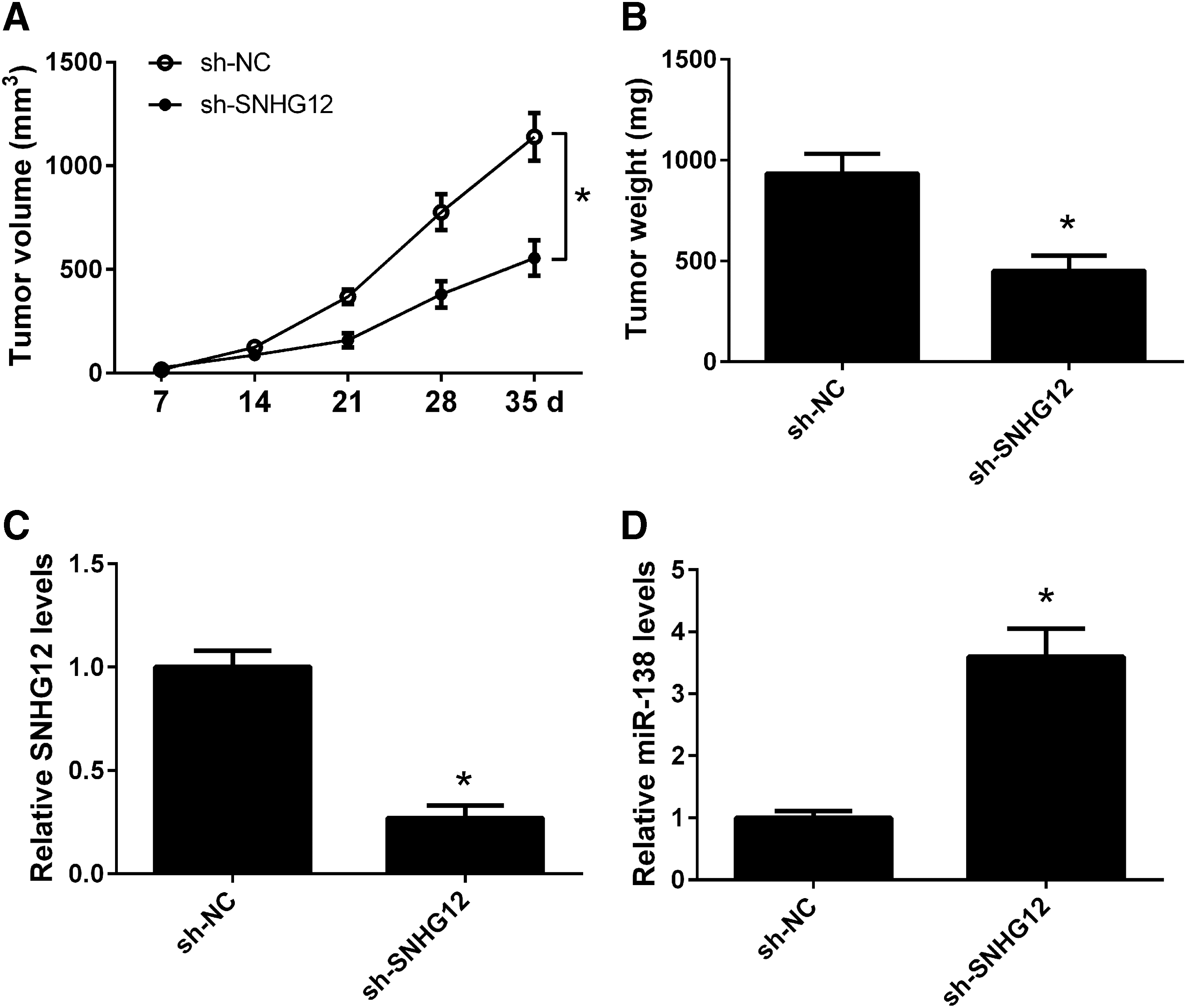
SNHG12 knockdown inhibited NSCLC tumor growth in a mouse model. H1299 cells transfected with sh-SNHG12 or sh-NC were subcutaneously injected into the right flank of the nude mice (n = 6)
Discussion
In this study, we found that SNHG12 was upregulated in NSCLC cell lines, compared with that in normal human bronchial epithelial cell line 16-HBE. Dual-luciferase reporter assay and RIP assay demonstrated that SNHG12 harbored the binding site of miR-138. Knockdown of SNHG12 inhibited growth and induced apoptosis of NSCLC cells via negatively regulating miR-138 expression. Knockdown of SNHG12 also inhibited xenograft tumor growth in a NSCLC mouse model.
SNHG12 was a novel lncRNA identified to be upregulated and function as an oncogene in several cancers. Zhai et al. (2015) first found that SNHG12 was significantly upregulated in endometrial cancer. SNHG12 knockdown suppressed proliferation and increased cell apoptosis and G1 phase arrest in endometrial cancer cells (Zhai et al., 2015). Ruan et al. (2016) found that SNHG12 expression was upregulated in osteosarcoma tissues and cells and knockdown of SNHG12 inhibited cell proliferation and migration via downregulating angiomotin gene expression. SNHG12 was also reported to be upregulated in triple-negative breast cancer tissues compared with normal tissues. Enforced expression of SNHG12 enhanced proliferation, migration, and reduced apoptosis of triple-negative breast cancer cells (Wang et al., 2017b). In hepatocellular carcinoma tissues, SNHG12 expression levels were significantly higher than those in adjacent normal tissues. SNHG12 acted as an endogenous sponge for miR-199a/b-5p to activate the NF-κB pathway, thus suppressing proliferation, inhibiting metastasis, and inducing apoptosis of hepatocellular carcinoma cells (Tian et al., 2017). A recent study suggested that SNHG12 expression levels were increased in colorectal cancer tissues and cells. SNHG12 knockdown inhibited proliferation, suppressed cell cycle, and induced apoptosis of colorectal cancer cells (Wang et al., 2017a). Consistent with these results, we, in the present study, found that SNHG12 was upregulated in NSCLC cells, and SNHG12 knockdown inhibited growth and induced apoptosis of NSCLC cells in vitro and suppress tumor growth of NSCLC cells in vivo.
The tumor suppressor miR-138 has been studied very extensively and deeply and the dysregulation of miR-138 is closely related to the development and progression of multiple cancers, including colorectal cancer (Long et al., 2013), pancreatic cancer (Tian et al., 2016), bladder cancer (Yang et al., 2016), esophageal squamous cell carcinoma (Gong et al., 2013), nasopharyngeal carcinoma (Liu et al., 2012), gallbladder carcinoma (Ma et al., 2015), head and neck squamous cell carcinoma (Jin et al., 2013), osteosarcoma (Zhu et al., 2016), ovarian cancer (Yeh et al., 2013), glioma (Wei et al., 2013), and NSCLC (Han et al., 2014). In this study, we found that SNHG12 directly interacted with miR-138 and regulated the miR-138 levels. Therefore, we deduced that the oncogenic function of SNHG12 depended on miR-138, or rather miR-138 target genes. Ye et al. (2015) reported that miR-138 inhibited NSCLC cell proliferation by targeting 3-phosphoinositide-dependent protein kinase-1 (PDK1). miR-138 inhibited proliferation, induced apoptosis and blocked G1/S transition of NSCLC cells in vitro and tumor growth of NSCLC cells in vivo through binding to the 3′-UTR of enhancer of zeste homolog 2 (EZH2) (Zhang et al., 2013). Cyclin D3 was also a target gene of miR-138 and upregulation of miR-138 inhibited proliferation, increased cisplatin sensitivity, and decreased migration of NSCLC cells by the interaction with cyclin D3 (Han et al., 2016). A recent study indicated that miR-138 suppressed cell proliferation and reversed epithelial–mesenchymal transition in NSCLC cells by targeting G-protein-coupled receptor kinase-interacting protein 1 (GIT1) and semaphorin 4C (SEMA4C), two oncogenes (Yang et al., 2016). miR-138 could also target yes-associated protein 1 (YAP1) oncogene, leading to repression of NSCLC cell growth (Xiao et al., 2016). Some miR-138 target genes, including sentrin/SUMO-specific protease 1 (SENP1) (Yang et al., 2014) and G protein-coupled receptor 124 (GPR124) (Gao et al., 2014), were involved in the radioresistance or chemoresistance of NSCLC cells.
In conclusion, our data suggested that SNHG12 knockdown inhibited cell growth and induced apoptosis by upregulating miR-138 in NSCLC and SNHG12 might be a potential target for gene therapy.
Footnotes
Disclosure Statement
No competing financial interests exist.