
Abstract
Intervertebral disc (IVD) degeneration is closely related to inflammatory cytokines, such as tumor necrosis factor alpha (TNF-α). The endoplasmic reticulum (ER) serves several important cell functions, which are essential for normal cell metabolism and survival. This study aims to clarify the role of ER stress and unfolded protein response (UPR) in TNF-α-induced biological changes in rat nucleus pulposus cells (NPCs) and IVD degeneration. In our research, rat NPCs were cultured with different concentrations of TNF-α in the presence or absence of ER stress inhibitors. Related genes and proteins were measured by immunofluorescence staining, quantitative real-time PCR, and Western blot analyses to monitor ER stress. Cell proliferation was evaluated by CCK-8 assay and cyclin D1 expression. Apoptosis was detected by flow cytometry and Western blot analyses. Our results showed that TNF-α induced the apoptosis of some NPCs in the early stage and then accelerated the proliferation of surviving cells. In addition, TNF-α stimulus upregulated ER stress markers and initiated UPR. However, these effects could be reversed by inhibitors, thereby reducing cell proliferation and enhancing apoptosis. In conclusion, ER stress reinforces the survival and proliferation of NPCs in TNF-α stimulus by activating UPR signaling, which could be an important therapeutic target in the future.
Introduction
L
As a pleiotropic pro-inflammatory cytokine and a trigger in IDD (Kang et al., 2015), tumor necrosis factor alpha (TNF-α) is highly expressed in degenerative IVD (Weiler et al., 2005). TNF-α amplifies inflammatory responses and induces NPC apoptosis and extracellular matrix (ECM) degradation, resulting in progressive IDD (Hiyama et al., 2013). However, our previous study proved that TNF-α enhances the proliferation of human NPCs through nuclear factor-κB (NF-κB), c-Jun N-terminal kinase (JNK), and p38 mitogen-activated protein kinase (p38 MAPK) pathways (Wang et al., 2015). Therefore, we assumed that different cell stress statuses lead to different biological phenomenon of apoptosis or proliferation of NPCs under TNF-α stimulus.
Endoplasmic reticulum (ER) is a vital organelle with biosynthetic capabilities. Accurate synthesis and folding of proteins by ER are essential for cell survival and normal functions (Kato et al., 2012). Endogenous and exogenous stresses may induce the stress response of ER (also called as ER stress) and further initiate unfolded protein response (UPR) (Fujimoto et al., 2016). UPR plays an important role in maintaining ER homeostasis. The three major ER stress branches include the following: inositol-requiring transmembrane kinase and endonuclease 1 (IRE1), PKR-like ER stress kinase (PERK), and activating transcription factor 6 (ATF6) (Ron and Walter, 2007).
Under normal conditions, these ER stress sensors are bound to glucose-regulated protein 78 (GRP78) and inactivated. When ER stress strikes, the GRP78 dissociates from these sensors and initiates downstream signaling pathways, thereby enhancing the protein folding capacity and accelerating the degradation of misfolded proteins (Boyce and Yuan, 2006). The activation of PERK leads to the phosphorylation of eukaryotic translation initiation factor 2α (eIF2α), which maintains cell stability and normal function (Harding et al., 2003). The activation of IRE1 cuts a 26-nucleotide intron from the mRNA of X-box binding protein 1 (XBP1), resulting in the production of spliced XBP1 (XBP1s) and the induction of ER chaperones that enhance ER folding capability (Acosta-Alvear et al., 2007; Ron and Hubbard, 2008). ATF6 is a transmembrane protein and located in the ER. Under ER stress, ATF6 is transported to Golgi apparatus, where it is cleaved to release the transcription factor (Shen et al., 2002).
There are extensive connections between inflammation and ER stress (Connor et al., 2012). Inflammation induced by lipopolysaccharide, TNF-α, IL-1 beta, and IL-6 can activate ER stress, and the stress caused by these inflammatory factors may be mediated by oxidative stress (reactive oxygen species [ROS]) (Nakajima and Kitamura, 2013). TNF-α can activate PERK- and IRE1-dependent pathways of UPR in a ROS-dependent manner. The UPR normally functions to prevent ROS accumulation and relieve stress (Xue et al., 2005). However, limited works have focused on the relationship between ER stress and TNF-α-induced NPC biological changes.
In view of the importance of UPR in cell function and survival, we hypothesized that ER stress is related to the cellular fates of proliferation or apoptosis in inflammatory microenvironment. TNF-α induces the apoptosis of some NPCs and also activates ER stress and promotes the survival and proliferation of NPCs through the UPR signaling pathway. In our experiments, we demonstrated that TNF-α could activate UPR, which is involved in ER stress. The inhibition of UPR weakened NPC proliferation and promoted apoptosis in the presence of TNF-α. Therefore, UPR played antiapoptotic and pro-survival roles in NPCs in response to TNF-α. Maintaining cellular UPR capability and stress status could be a potential and novel therapeutic strategy for IVD regeneration.
Materials and Methods
Isolation and culture of rat NPCs
Animal experiments were performed according to the protocol approved by the Institutional Animal Care and Use Committee of Southeast University (Nanjing, China).
Sprague-Dawley rats (male, n = 20) were euthanized by carbon dioxide (CO2). Rat NP tissues were collected as previous studies (Hiyama et al., 2010), resuspended in DMEM/F12 containing 10% FBS and antibiotics, and incubated at 37°C with 5% CO2 in a humidified incubator. The medium was changed every 3 days. The passage three cells were used in the following studies.
Cell proliferation and cytotoxicity assay
NPCs were transferred into 96-well plates at a density of 2000 cells/well and treated with gradient concentrations of TNF-α (PeproTech), 4-phenylbutyrate (4-PBA) (Sigma), GSK2606414 (ApexBio), and 4μ8C (ApexBio). The cells were then cultured at 37°C in 5% CO2 air atmosphere from 24 to 96 h. Cell proliferation was quantified using CCK-8 solution (KeyGen, China) by the absorbance at 450 nm by Multiskan MK3 (Thermo Scientific). For inhibitor treatment experiments, NPCs were pretreated with inhibitors for 4-PBA (1 mM), GSK2606414 (PERKi, 2 μM), and 4μ8C (IRE1i, 10 μM) for 24 h before TNF-α (10 ng/mL) treatment.
Apoptotic analyses by flow cytometry
Apoptosis was evaluated by flow cytometry using the annexin V-FITC/PI Apoptosis Detection Kit (KeyGen) according to the manufacturer's instructions. After washing twice with phosphate-buffered saline (PBS), floating and attached cells were both collected and resuspended in buffer containing annexin V-FITC and PI. Cells were quantified by flow cytometry (BD Accuri C6). The results are expressed as the proportions of apoptotic cells in each group.
RNA isolation and quantitative real-time PCR
NPCs were collected, and total mRNA was isolated using TRIzol reagent (Invitrogen). Reverse transcription was performed using PrimeScript™ RT Master Mix (RR036A; TaKaRa, Dalian, China) following the manufacturer's instructions. The primer sequences are presented in Table 1. Real-time PCR (RT-PCR) analysis was performed using SYBR Premix EX Taq™ (RR420A; TaKaRa) in StepOnePlus RT-PCR system (Applied Biosystems) according to the manufacturer's instructions. Relative gene expression was measured using the 2–ΔΔCt method with β-actin gene as internal control.
eIF2α, eukaryotic translation initiation factor 2α; GRP78, glucose-regulated protein 78; qRT-PCR, quantitative real time PCR; XBP1, X-box binding protein 1.
Protein extraction and Western blot analysis
Total protein was extracted using whole-cell lysis assay (KeyGen). Protein concentrations were determined by BCA assay (Beyotime, China). Proteins were separated using 10% sodium dodecyl sulfate–polyacrylamide gel electrophoresis and transferred onto polyvinylidene fluoride membranes (Millipore). The membranes were blocked with 5% skimmed milk in TBST for 1.5 h and incubated overnight at 4°C with primary antibodies (Table 2). The membranes were then incubated with secondary antibodies (1:5000, ab6721; Abcam) diluted in 5% bovine serum albumin (BSA)/TBST for 2 h at room temperature. After detection, bands were assessed by densitometric analysis. Protein expression level was normalized to that of vinculin or β-actin.
Immunofluorescence staining
Cultured NPCs were treated with TNF-α (0, 5, 10, and 20 ng/mL) for 24 h, fixed with 4% paraformaldehyde for 25 min, permeabilized with 0.3% Triton X-100 for 20 min, and blocked with 5% BSA for 30 min at room temperature. The cells were then incubated with rabbit anti-GRP78 (1:400, ab21685; Abcam) at 4°C overnight, followed by Alexa Fluor 647 goat anti-rabbit IgG (1:1000, ab150079; Abcam) for 1 h at 37°C. Nuclei were counterstained with 4′6-diamidino-2-phenylindole (DAPI; Sigma-Aldrich, Ireland) and observed on a fluorescence microscope (Olympus, Japan).
Statistical analyses
Data are presented as mean ± SD. Statistical analyses were carried out using GraphPad Prism 6 (GraphPad Software, Inc.). Differences among groups were evaluated using unpaired Student's t-test. p-values <0.05 were considered significant.
Results
TNF-α regulated the apoptosis and proliferation of NPCs
Rat NPCs were treated with gradient concentration of TNF-α to investigate the biological effects of TNF-α on the apoptosis and proliferation of NPCs. Flow cytometry analysis and CCK-8 assay were performed to calculate the rate of cell apoptosis and proliferation. In addition, the mRNA and protein expression levels of cyclin D1 were measured by quantitative RT-PCR (qRT-PCR) and Western blot analyses, respectively. At TNF-α concentrations higher than 10 ng/mL, the proportion of apoptotic cells was increased by TNF-α treatment for 24 h (Fig. 1A–C). After 24 h, the proliferation rate of NPCs increased upon exposure to TNF-α (5, 10, and 20 ng/mL) stimulus (Fig. 1D). In addition, cyclin D1 expression was significantly increased at the mRNA and protein levels by the gradient concentration of TNF-α after 24 h. This finding indicates the rapid proliferation of surviving cells (Fig. 1E–G). Hence, TNF-α induced the apoptosis of some NPCs and accelerated the proliferation of surviving cells.

Effects of TNF-α on the apoptosis and proliferation of rat NPCs.
TNF-α activated ER stress and UPR signaling in rat NPCs
Different concentrations of TNF-α (from 5 to 20 ng/mL) were used to culture rat NPCs and demonstrate the role of TNF-α in regulating ER stress and UPR signaling. The expression levels of ER stress markers and UPR proteins were analyzed. Based on immunofluorescence staining, the expression of the ER stress marker GRP78 was higher in NPCs upon TNF-α exposure than that in the normal group (Fig. 2A, B). The mRNA (Fig. 2C–F) and protein (Fig. 3A–C) expression levels of ER stress-related genes were elevated significantly by TNF-α stimulus at 24 h. We also assessed the expression of phosphorylated eIF2α (p-eIF2α) and XBP1s by Western blot, which represented the activation status of UPR signaling. The results also indicated the upregulated trend of activation (Fig. 3D–F). After continuous TNF-α (10 ng/mL) treatment for 96 h, the expression levels of p-eIF2α and XBP1s were increased compared to control at 24, 48, 72, and 96 h (Fig. 3G–I). Hence, TNF-α induced ER stress and activated UPR signaling pathways, including those involving PERK/eIF2α and IRE1/XBP1 in rat NPCs.
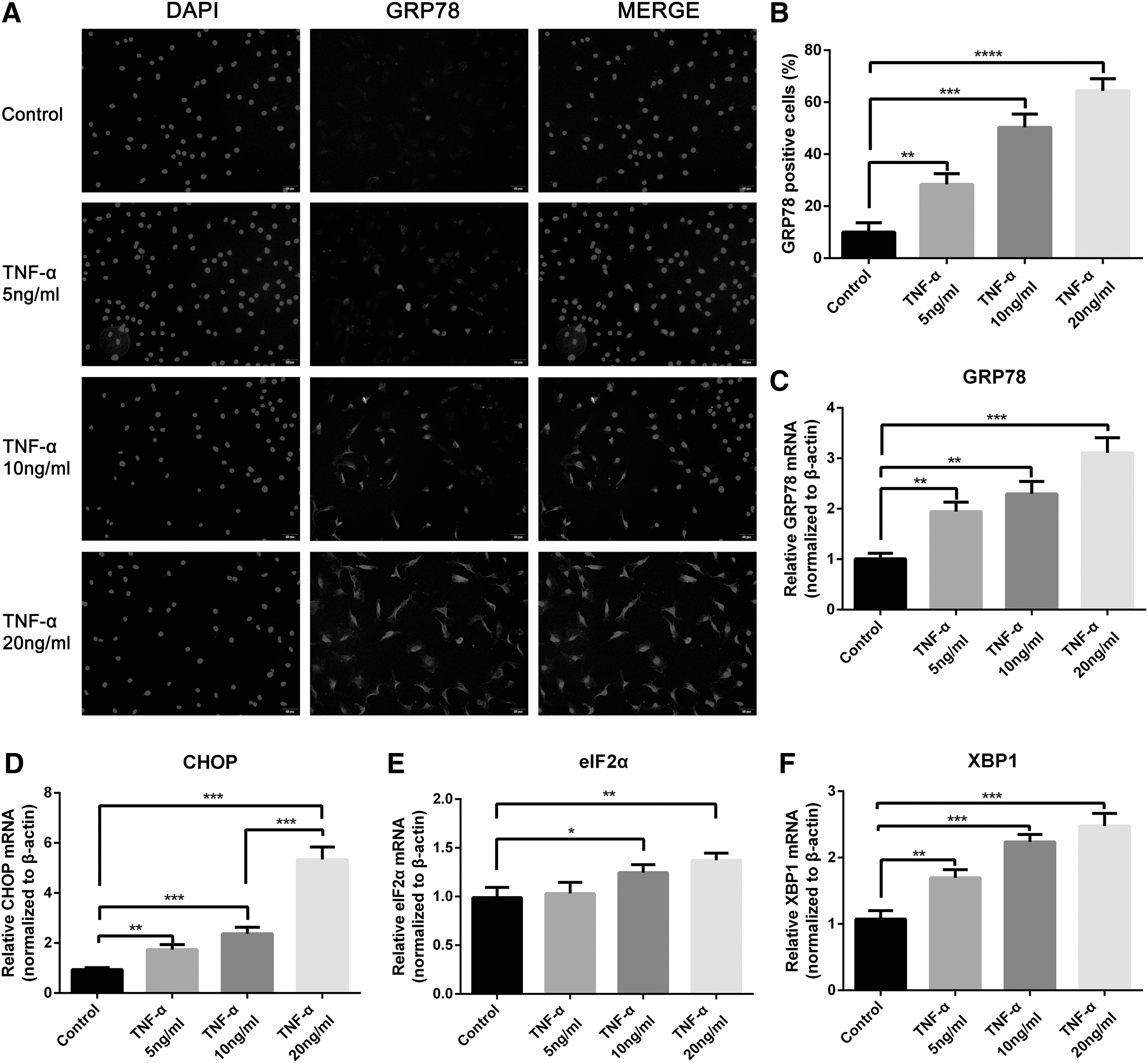
TNF-α activated ER stress in rat NPCs. Cells were treated with different concentrations of TNF-α (0, 5, 10, and 20 ng/mL) for 24 h.

TNF-α activated PERK/eIF2α and IRE1/XBP1 pathways of UPR signaling in rat NPCs. Cells were treated with gradient concentrations of TNF-α (0, 5, 10, and 20 ng/mL) for 24 h.
ER stress promoted the survival and proliferation of NPCs under TNF-α stimulus
Rat NPCs were exposed to 10 ng/mL TNF-α with or without pretreatment with the ER stress inhibitor 4-PBA for 24 h. First, the cytotoxicity of 4-PBA was detected by flow cytometry and CCK-8 assay. Within 24 h, low concentrations (0.5 and 1 mM) exerted no significant effect on cell survival; however, 4-PBA concentration higher than 2 mM enhanced apoptosis (Fig. 4A–C). Subsequently, we pretreated NPCs with 1 mM 4-PBA for 24 h and exposed to 10 ng/mL TNF-α for another 24 h. Inhibition was verified by decreased expression levels of proteins associated with ER stress and UPR (Fig. 4D–I). Flow cytometry analyses confirmed that the ER stress inhibitor promoted the induction of apoptosis triggered by TNF-α (Fig. 5A, B). Moreover, the Western blot revealed that the inhibition of ER stress increased the expression levels of apoptosis-related proteins, namely, cleaved-caspase3 and Bax, and decreased those of the antiapoptosis-related protein Bcl2 (Fig. 5C–F). These results indicated that the inhibited ER stress sensitized NPCs to TNF-α-mediated apoptosis. In addition, cell proliferation rate by CCK-8 assay (Fig. 5G) and cyclin D1 expression by Western blot analysis (Fig. 5H, I) decreased significantly upon pretreatment with 4-PBA. Hence, the ER stress inhibitor weakened cell proliferation induced by TNF-α. We conclude that ER stress promoted the survival and proliferation of NPCs under TNF-α stimulus.
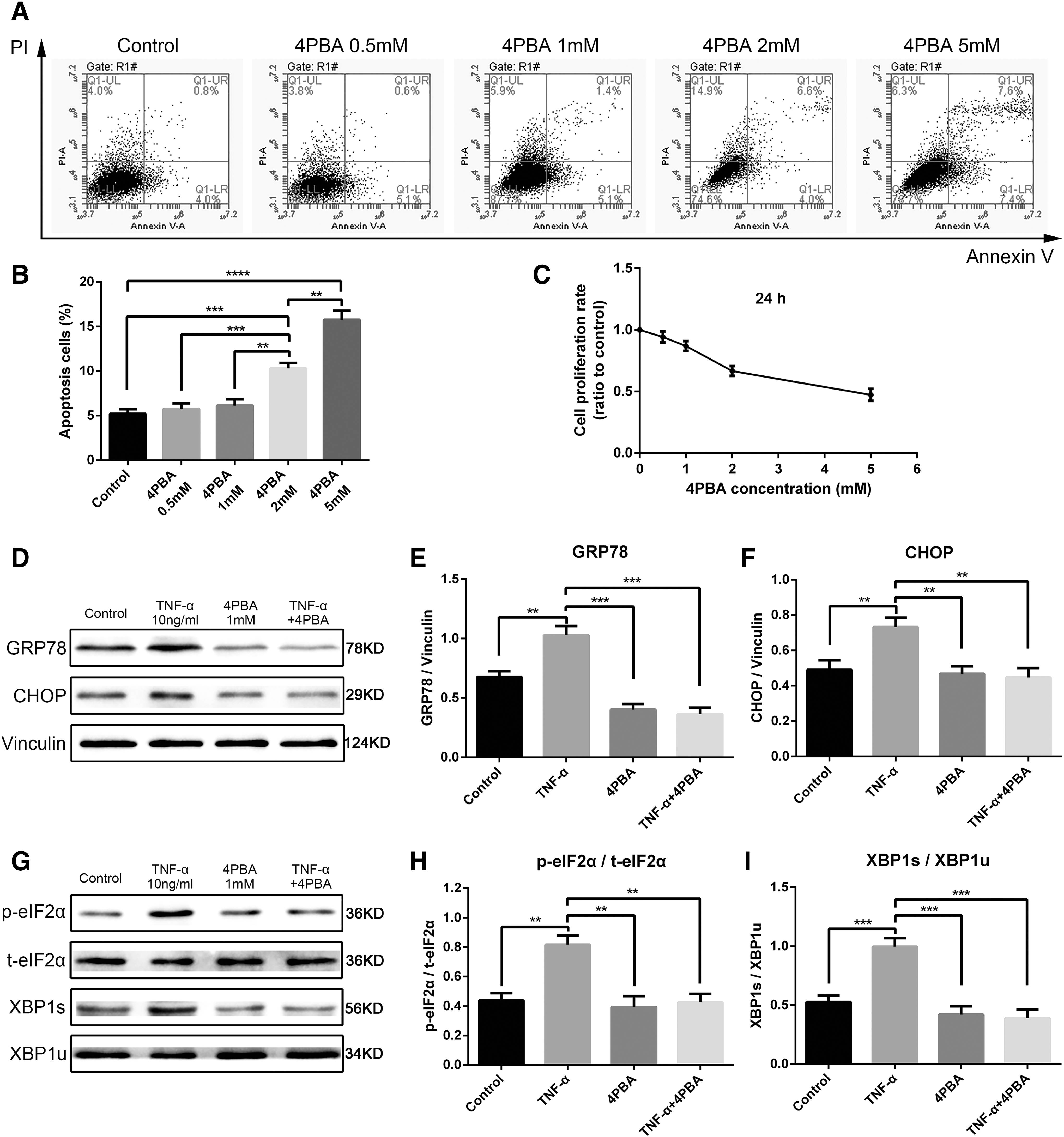
4-PBA-related cytotoxicity detection and its inhibition of ER stress.
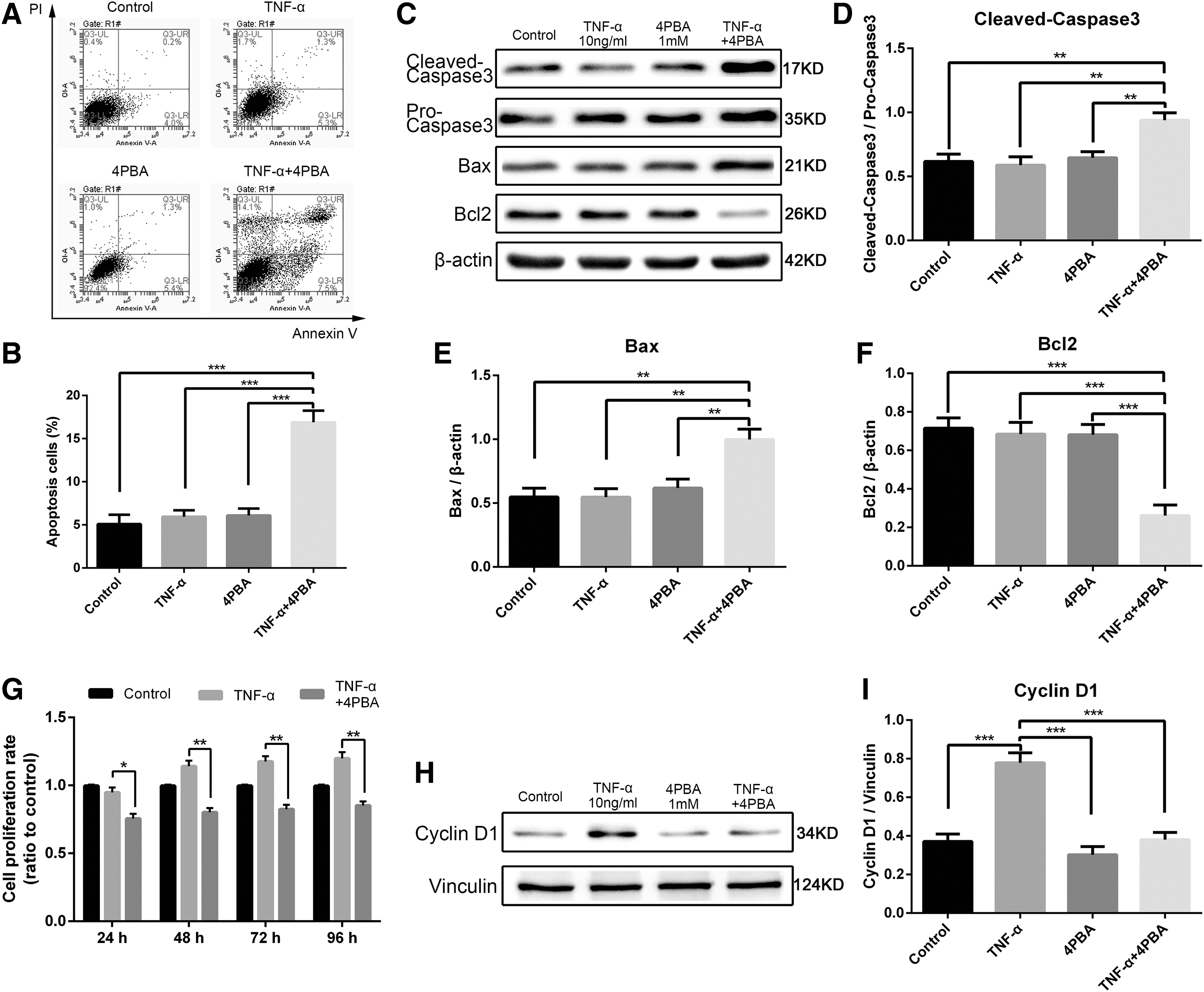
Role of ER stress in rat NPC biological changes induced by TNF-α. Cells were exposed to 10 ng/mL TNF-α with or without pretreatment with 1 mM chemical chaperone 4-PBA for 24 h.
ER stress promoted the survival and proliferation of NPCs upon exposure to TNF-α stimulus through the UPR pathway
The specific PERK inhibitor GSK2606414 (PERKi) and the IRE1 inhibitor 4μ8C (IRE1i) were used to block PERK and IRE1 pathways, respectively, and elucidate the role of their corresponding pathways in regulating cell apoptosis or proliferation induced by TNF-α. First, concentrations that do not lead to cytotoxicity were identified by CCK-8 assay (Fig. 6A, B). Within 24 h, 2 μM PERKi and 10 μM IRE1i had no significant effect on cell survival. Furthermore, we pretreated cells with or without 2 μM PERKi and10 μM IRE1i for 24 h before exposure to 10 ng/mL TNF-α. The expression levels of p-eIF2α and XBP1s were assessed by Western blot analysis after 24 h (Fig. 6C, D). The TNF-α-mediated induction of UPR pathway proteins, namely, p-eIF2α and XBP1s, were significantly downregulated by PERKi and IRE1i, respectively (Fig. 6E, F). Flow cytometry analysis was performed to detect the proportion of apoptotic cells. Both PERKi and IRE1i increased cell apoptosis induced by TNF-α (Fig. 7A–D). In addition, CCK-8 assay (Fig. 7E, F) and Western blot analyses (Fig. 7G–J) both proved that the proliferation of NPCs weakened upon exposure to the UPR inhibitor. Hence, PERK/eIF2α and IRE1/XBP1 pathways regulated UPR-mediated survival and proliferation of NPCs under TNF-α stimulus.
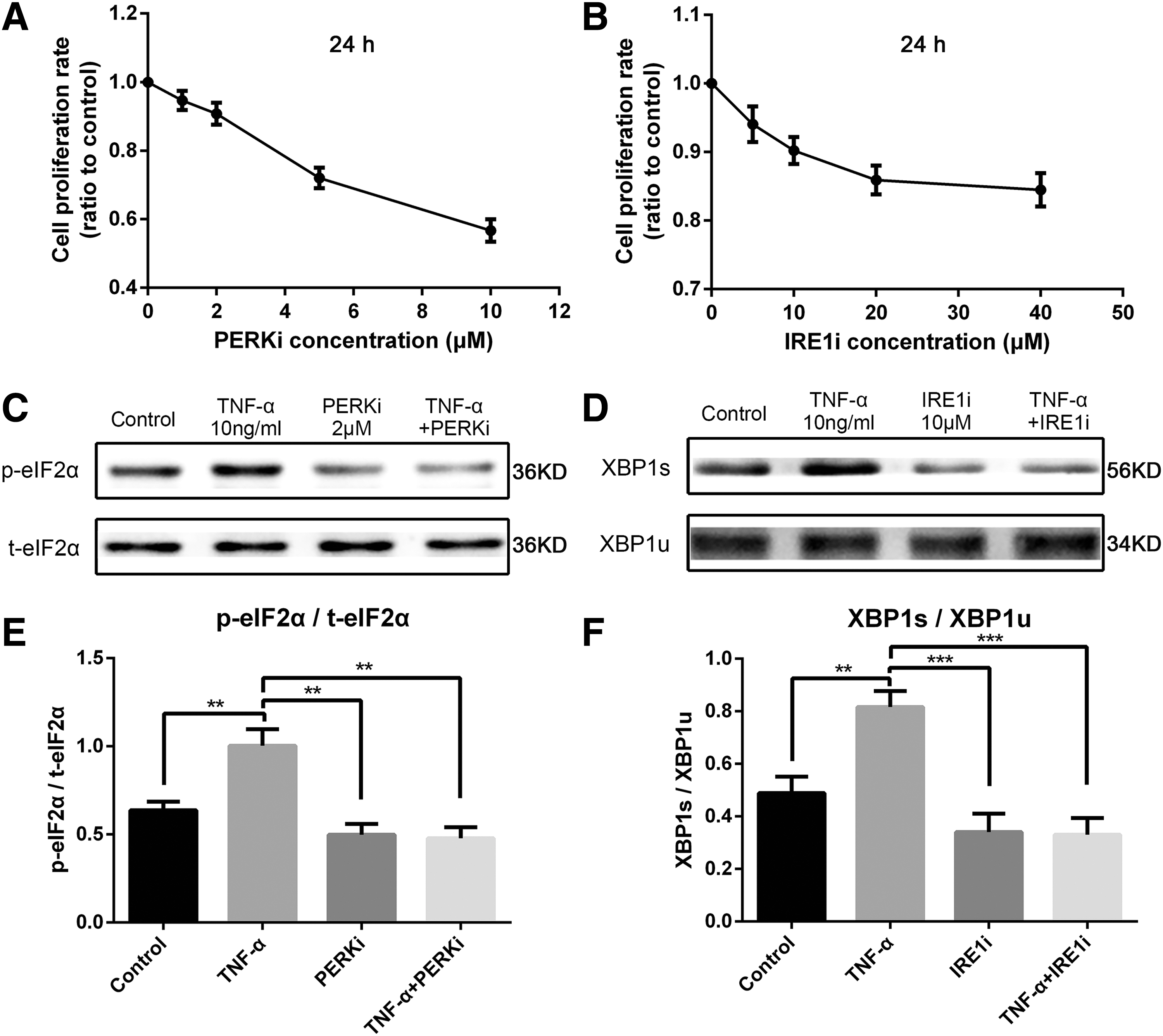
PERKi- and IRE1i-related cytotoxicity and their ability to inhibit UPR pathways.
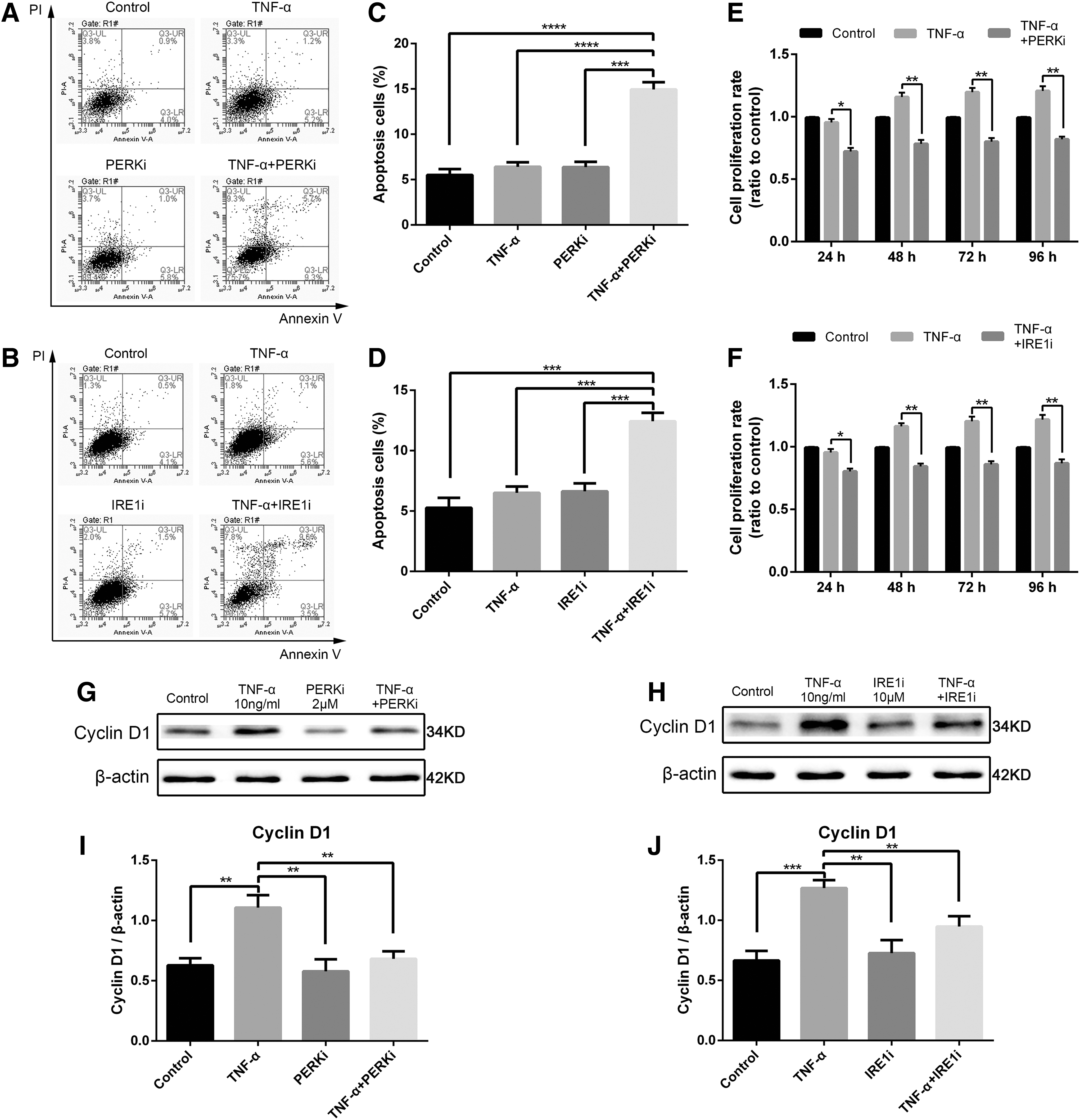
Role of UPR in rat NPC apoptosis and proliferation induced by TNF-α. Cells were exposed to 10 ng/mL TNF-α with or without 2 μM PERKi and 10 μM IRE1i pretreatment for 24 h, respectively.
Discussion
Cellular loss and the following ECM decrease play a crucial role in the progression of IDD. However, the mechanism of NPC apoptosis in the degeneration of IVD remains unclear. Accumulating evidence confirms that TNF-α significantly increased in degenerative IVD and promoted NPC loss (Purmessur et al., 2013). Nevertheless, in our observation, the proliferation of rat NPCs significantly increased following treatment with TNF-α for 24 h. In our previous studies, we first demonstrated that ER stress participated in IDD (Xie et al., 2017). And to the best of our knowledge, it was also the first time that we connected ER stress with TNF-α in the apoptosis and proliferation of NPCs.
ER stress and the UPR are related to main inflammatory and stress-signaling networks through several mechanisms (Chaudhari et al., 2014). Our research proved that the inflammatory factor TNF-α induces ER stress and UPR by increasing the expression of markers, including GRP78, downstream expression of CHOP, PERK-mediated p-eIF2α, and IRE1-mediated induction of XBP1s. ER responds to stress by initiating UPR to restore normal ER homeostasis and elicit cytoprotective effects. In addition, ER stress is related to cell proliferation. Some scholars have confirmed that ER stress mediates the induction of miR-3648 in human cells, thereby downregulating the expression of the negative regulator of Wnt signaling to increase cell proliferation (Rashid et al., 2017). GRP78 regulates cell cycle-associated proteins through the Akt pathway (Lee et al., 2017). However, when prolonged and severe ER stress exceeds the capacity of UPR, the ER homeostasis cannot be restored, and cells with disrupted ER functions undergo apoptosis (Oyadomari and Mori, 2004; Yoo et al., 2012; Ariyasu et al., 2017). Thus, we hypothesize that ER stress activates the cytoprotective UPR pathway to promote cell survival and cell proliferation during NPC apoptosis induced by TNF-α.
As a chemical chaperone, 4-PBA can repress ER stress and UPR activation by interrupting the recognition of the ER monitor of unfolded or misfolded proteins (Inden et al., 2007; Haberzettl et al., 2009). Our previous studies demonstrated that when ER stress was blocked by 4-PBA, NPCs under acid stimulus showed high apoptotic rate; hence, ER stress is a key factor leading to the acid resistance of NPCs and attenuated acid-induced apoptosis of NPCs (Xie et al., 2017). In the present study, TNF-α stimulus combined with 4-PBA pretreatment downregulated ER stress-related protein expression, increased cell apoptosis rate, and significantly decreased cell proliferation.
However, some scholars have reported that 4-PBA could protect cells from apoptosis by attenuating ER stress (Zamarbide et al., 2013), whereas in our model 4-PBA does not protect cells against TNF-α even though it suppresses ER stress. The reasons may be as follows: First, as a chemical chaperone, 4-PBA bound to unfolded proteins and competitively inhibited the binding of GRP78. The unfolded proteins are not recognized by ER chaperone GRP78; therefore, the UPR is not activated and the downstream cell protection mechanisms cannot work normally. Consequently, cell apoptosis increases in the presence of intense and prolonged external stimulation of TNF-α. Second, NPC is a special kind of cell, which living in the largest avascular structure in the body is different from other cell types. The microenvironment of ischemic and anoxia in IVD makes NPCs usually being stressful, and ER stress is easy to induce in NPCs. Moreover, NPCs are required with abilities of stronger ECM synthesis and accurate protein synthesis. All of our studies suggested that ER stress mainly plays cell protective role in NPCs and apoptosis pathway activated under severe stress (Supplementary Fig. S1; Supplementary Data are available online at
ER stress leads to the motivation of the three branches (PERK, IRE1, and ATF6) of UPR; of which, we focused on the role of PERK and IRE1-induced arms. The inhibition of PERK or IRE1 obtained the same results as that of 4-PBA pretreatment. All these results indicated that ER stress facilitated the survival and proliferation of NPCs by activating UPR signaling.
The PERK branch of UPR is strongly protective at moderate levels of ER stress. PERK signaling through eIF2α is vital for UPR and stress tolerance (Harding et al., 2000). This signaling is critical for survival and inhibiting it induced cells to become more sensitive to ER stress induced by TNF-α. At moderate concentrations of TNF-α, the proportion of apoptotic cells significantly increased, and cell proliferation decreased. The main protective mechanisms may be described as follows: PERK promotes cell survival during ER stress through inhibition of protein synthesis, thereby preventing the accumulation of misfolded or unfolded proteins (Cojocari et al., 2013). During prolonged ER stress, PERK induces autophagy (Kania et al., 2015).
ER stress also triggers the IRE1-dependent pathway. Activated IRE1 cuts out 26 nucleotides of the unspliced XBP1 (XBP1u) mRNA to generate the XBP1s mRNA, which can be translated to XBP1s (Hosogai et al., 2007). XBP1s is a strategic regulator of UPR because it initiates genes that are responsible for restoring the ER folding capacity (Acosta-Alvear et al., 2007). 4μ8C is an effective inhibitor of the splicing activity in response to ER stress (Cojocari et al., 2013), thereby suppressing TNF-α-induced XBP1 splicing and downstream gene expression. IRE1/XBP1 signaling pathway is a part of the cellular program that controls the development and survival of immune cells against stress (Tavernier et al., 2017). Based on the flow cytometry results of NPCs, TNF-α-induced apoptosis was considerably increased by 4μ8C, resulting in a drastic decrease in cyclin D1 protein levels. These results are in agreement with relevant theories indicating that IRE1 sensor is generally associated with pro-survival and proliferation pathways in response to ER stress by regulating multiple chaperones.
Overall, the activation of UPR enhanced the survival and proliferation of rat NPCs through PERK/eIF2α and IRE1/XBP1 pathways. Hence, UPR played a crucial role in promoting tolerance to TNF-α-induced stress (Fig. 8A, B).

Schematic of ER stress involved in TNF-α-induced NPC biological changes through UPR pathways.
Conclusions and Perspectives
Our experiments proved that TNF-α induced the apoptosis of rat NPCs and the proliferation of surviving cells by activating ER stress and UPR signaling. Pretreatment with ER stress and UPR pathway inhibitors weakened cell proliferation and promoted the apoptosis of NPCs induced by TNF-α. Hence, UPR played antiapoptotic and pro-survival roles in TNF-α induced biological changes in NPCs. Moreover, UPR enhanced the cell survival and proliferation rates in response to inflammation stimulus and contributed to protection against TNF-α.
ER dysfunction is an important factor in a wide range of diseases (Hetz et al., 2013). As discussed in this article, therapeutic strategies for promoting cell survival may reinforce protective UPR signaling responses involved in adaptation to stress, thereby reducing TNF-α-induced apoptosis of NPCs during IVD degeneration.
Footnotes
Acknowledgments
The research was supported by grants from the National Natural Science Foundation of China (81572170, 81572190, 81702203) and the Fundamental Research Funds for the Central Universities and Postgraduate Research & Practice Innovation Program of Jiangsu Province (KYCX17_0182, KYLX16_0302, 2242016K40149, 2242017K3DN06).
Disclosure Statement
No competing financial interests exist.
References
Supplementary Material
Please find the following supplemental material available below.
For Open Access articles published under a Creative Commons License, all supplemental material carries the same license as the article it is associated with.
For non-Open Access articles published, all supplemental material carries a non-exclusive license, and permission requests for re-use of supplemental material or any part of supplemental material shall be sent directly to the copyright owner as specified in the copyright notice associated with the article.