
Abstract
Diabetes is a worldwide health problem with increasing prevalence. Some reports indicate the interplay between bone and glucose metabolism. The imbalance between bone resorption and formation resulted in the structural integrity and strength of bone. Glucagon-like peptide-1 (GLP-1) and its agonists (Liraglutide) have an anabolic action on bone remodeling by stimulating osteoblast differentiation as well as increasing osteoblast longevity. However, the underlying mechanisms remain elusive. We detected the presence of GLP-1 receptor (GLP-1R) in MC3T3-E1 cells via immunocytochemistry assay. Alkaline phosphatase activity assay, alizarin red stain, quantitative real-time polymerase chain reaction, and western blot were employed to detect the effect of Liraglutide on osteogenic differentiation. Liraglutide promoted the expression of GLP-1R in a dosage- and time-dependent manner, and it enhanced the osteogenic differentiation in MC3T3-E1 cells. Liraglutide application improved the levels of Smad2/3 and p-Smad2/3; however, the silencing of Smad2/3 blocked the osteogenic differentiation induced by Liraglutide. What is more, the application of PI3K and Wnt inhibitors inhibited the upregulation of Akt, p-Akt, β-catenin, Smad2/3, and p-Smad2/3 induced by Liraglutide. Liraglutide facilitated the osteogenic differentiation via the regulation of Smad2/3 via PI3K/AKT and Wnt/β-catenin pathways. These data revealed a new mechanism of Liraglutide inducing osteogenic differentiation and provided theory evidence to maintain normal bone metabolism during diabetes therapy.
Introduction
D
Incretin hormone is a kind of peptide that is secreted in the gastrointestinal tract in response to nutrient intake and it potentiates insulin release in a glucose-dependent manner (Nauck and Meier, 2018). Some evidence showed that incretin hormones have an anabolic action on bone remodeling by stimulating osteoblast differentiation as well as increasing osteoblast longevity (Luo, et al., 2016; Ramsey and Isales, 2017). Glucagon-like peptide-1 (GLP-1) is a key incretin hormone secreted from gut L cells. GLP-1 stimulates insulin secretion by pancreatic β cells in a glucose-dependent way and suppresses glucagon release from α cells (Kazafeos, 2011). GLP-1 exerts its activities via the interaction with GLP-1 receptor (GLP-1R), which is widely distributed in bone (Campbell and Drucker, 2013). However, GLP is rapidly degraded and inactivated by the ubiquitous protease dipeptidyl peptidase IV (DPP-IV) in the circulation. So GLP-1R agonists (GLP-1RA) are synthesized for clinical use due to an extended half-life with their resistance to degradation by DPP-IV (Ryan et al., 2011). Liraglutide belongs to a synthetic GLP-1RA with a prolonged half-life (Coulter et al., 2018). Liraglutide shares 97% homology with the structure of human GLP-1 (Gallwitz, 2015). It is synthesized on the basis of a GLP-1 backbone, where arginine replaces lysine 34 and a palmitoyl fatty acid is added at position 26 via a glutamic acid spacer (Russell-Jones, 2009). Liraglutide is widely used in the treatment of diabetes (Lambadiari et al., 2018) and obesity to decrease weight gain (Camkurt et al., 2018). What is more, Liraglutide is reported to relieve cardiac dilated function (Boyle et al., 2018) and reduce oxidative stress (Rizzo et al., 2015) in diabetic patients.
Previous research reveals that Liraglutide is involved in facilitating bone formation and suppressing bone resorption. In diabetic Goto-Kakizaki rats (Sun et al., 2015), type 1 diabetic mice (Mansur et al., 2015), ovariectomized rats (Lu et al., 2015), and type 2 diabetes and obesity (Iepsen et al., 2015), Liraglutide significantly prevents deterioration of the quality of the bone matrix. Although some studies have been performed to determine the beneficial anabolic effects of Liraglutide on skeletal health, the downstream molecular mechanisms underlying the osteogenic effects still remain elusive. It was reported that Liraglutide facilitates osteogenic proliferation and differentiation of MC3T3-E1 cells via PI3K/AKT, ERK1/2, and cAMP/PKA pathways (Wu et al., 2017). It is also reported that Liraglutide attenuates the osteoblastic differentiation through AMPK/mTOR signaling in MC3T3-E1 cells (Hu et al., 2016). Further, Liraglutide represses serum deprivation-induced apoptosis in MC3T3-E1 cells through cAMP/PKA/β-catenin and PI3K/AKT/GSK3β pathways (Wu et al., 2018).
Transforming growth factor-β (TGF-β) is directly or indirectly involved in a variety of cellular functions, including cell proliferation, differentiation, and apoptosis (Yan et al., 2018). Smad proteins are the major mediators of the TGF-β pathway to regulate the transcription of their targets (Chen et al., 2018). Smad2/3, as critical components of TGF-β signaling, are closely associated with the regulation of bone formation (de Kroon et al., 2017). In our study, we aimed at exploring the relationship between Liraglutide and the TGF-β/Smad pathway to reveal the new mechanism of Liraglutide functioning. During the study, we found that Liraglutide regulated GLP-1R expression in a dosage- and time-dependent manner in MC3T3-E1 cells. We also found that Liraglutide modulated an increase in the alkaline phosphatase (ALP) activity and bone matrix mineralization, and it promoted the expression of osteogenic markers runt-related transcription factor-2 (RUNX2) and osteocalcin (OCN). Liraglutide enhanced the levels of Smad2/3, p-Smad2/Smad2, and p-Smad3/Smad3; however, the silencing of Smad2/3 blocked the osteogenic differentiation induced by Liraglutide. The inhibitors of PI3K and Wnt suppressed the expression of Smad2/3 and β-catenin induced by Liraglutide. Taken together, these data revealed that Liraglutide was involved in the osteogenic differentiation via regulating the expression of Smad2/3, and Liraglutide modulated the levels of Smad2/3 through PI3K/AKT and Wnt/β-catenin pathways. These findings provided new evidence for the essential function of Liragultide in bone metabolism therapy.
Materials and Methods
Cell culture
Osteocytic cell line MC3T3-E1 was purchased from Yingniurui Biological Medicine Technology Co Ltd. (Wuxi, Jiangsu, China). MC3T3-E1 cells were cultured with alpha minimal essential medium (α-MEM; Hyclone) supplemented with 10% fetal bovine serum (FBS; Hyclone), 2 mM L-glutamine, 100 U/mL penicillin, and 100 μg/mL streptomycin at 37°C in a humidified incubator of 5% CO2. The culture medium was changed every 2–3 days.
Cell proliferation ability detection by cell counting kit 8
MC3T3-E1 cells were seeded into a 96-well plate at the density of 2 × 103 cells/well. After 24, 48, and 72 h, 10 μL of cell counting kit 8 (CCK8) stock solution were added to each well at each time point, and the plates were further incubated for 4 h at 37°C. The OD values were detected at the wavelength of 450 nm in a microplate reader (Thermo). Survival rate (%) = ODexperimental group/ODcontrol group × 100%.
Cell cycle distribution analyzed by flow cytometry
MC3T3-E1 cells were seeded into a six-well plate at 5 × 106 cells/well. After 24 h, cells were cultured with 0, 10−9, 10−8, or 10−7 M Liraglutide. The control group was administered with the same volume of physiological saline. After 72 h of incubation, cells were washed three times with phosphate-buffered saline (PBS). After digestion and centrifugation, the cells were resuspended with 0.5 mL PBS and treated with 3.5 mL 70% pre-cooling ethanol overnight at 4°C. The next day, the cells were washed three times with PBS, and they were treated with 1 mL of propidium iodide (KeyGEN Biotech, Nanjing, China) containing 0.2 mg of RNase A for 15 min at 37°C. Cell cycle was tested by a flow cytometer (BectonDickinson).
Immunocytochemistry
MC3T3-E1 cells were seeded into a six-well plate with cover slips at 1 × 104 cells/well. After 24 h, the cells were cultured with osteogenic medium containing 0, 10−9, 10−8, or 10−7 M Liraglutide. The control group was administered with the same volume of physiological saline. After 48 h of incubation, the cells were fixed with 4% paraformaldehyde for 30 min, and they were permeabilized with 0.3% Triton X-100 in PBS at room temperature for 10 min. After being washed with PBS, the cells were blocked with 5% bovine serum albumin for 30 min at 37°C. Then, the cells were incubated with rabbit polyclonal anti-GLP-1R antibody (ABclonal Biotechnology Co., Ltd.; 1:500 dilution) at 4°C overnight. The next morning, after being washed with PBS, the cells were probed with Alexa Fluor 488-labeled sheep anti-rabbit IgG (Invitrogen; 1:10,000 dilution) for 2 h at room temperature. After PBS washes, the nuclear was stained with 4′,6′-diamino-2-phenylindole (DAPI, 1 mg/mL; Sigma) for 10 min at room temperature. Images from five non-overlapping fields were captured by using an inverted laser confocal microscope (Leica, Germany).
Quantitative real-time polymerase chain reaction
MC3T3-E1 cells were seeded into six-well plates. After 24 h of incubation, the cells were treated with osteogenic medium containing 0, 10−9, 10−8, or 10−7 M Liraglutide for 3, 7, 14, and 21 days, respectively. The control group was incubated with the α-MEM supplemented with 10% FBS. Total RNA was extracted with TRIzol reagent (CWBIOtech, Beijing, China). RNA was reverse transcribed with GoTaq®Reverse Transcription System (Promega, Madison, WI). Polymerase chain reaction (PCR) was performed with GoTaq®Green Master Mix (Promega) in a Light Cycler instrument (BioRad). The relative expression of target genes was normalized to GAPDH with the 2−ΔΔCT method (Schmittgen and Livak, 2008). The primers for quantitative real-time PCR (qRT-PCR) were listed as follows: GLP-1R forward primer: 5′-tctccaaactgaaggctaat-3′, reverse primer: 5′-gtccatcacaaaggcaaa-3′; RUNX2 forward primer: 5′-ggactgggtatggtttgtat-3′, reverse primer: 5′-gctgaagaggctgtttga-3′; OCN forward primer: 5′-accacatcggctttcagg-3′, reverse primer: 5′-catagggctgggaggtca-3′; GAPDH forward primer: 5′-gcaagttcaacggcacag-3′, reverse primer: 5′-cgccagtagactccacgac-3′.
Western blot
MC3T3-E1 cells at logarithmic phase were seeded into six-well plates. After 24 h, the cells were cultured with osteogenic medium containing 0, 10−9, 10−8, or 10−7 M Liraglutide for 3, 7, 14, and 21 days, respectively. The control group was incubated with the α-MEM supplemented with 10% FBS. Proteins were extracted by using RIPA buffer (Sigma). Protein concentration was determined by BCA kit (Beyotime Biotechnology, Shanghai, China). Proteins were subjected to SDS-PAGE and transferred onto polyvinylidene fluoride membranes (Millipore, Bedford, MA). After being blocked with 5% bovine serum albumin for 1 h at room temperature, the membranes were incubated with rabbit anti-GLP-1R (1:500), anti-Runx2 (1:500), anti-OCN (1:500), anti-GAPDH (1:1000) (ABclonal Biotechnology Co., Ltd.), anti-Smad2 (1:500), anti-Smad3 (1:500), anti-p-Smad2 (1:300), or anti-p-Smad3 (1:300) (Abcam) antibodies at 4°C overnight, respectively. The next day, membranes were washed with TBST and probed with the corresponding horseradish peroxidase conjugated goat anti-rabbit immunoglobulin G secondary antibody (1:5000; Abcam) for 2 h at room temperature. After TBST washes, membranes were developed with an enhanced chemiluminescence kit (PerkinElmer). The relative expression of target proteins was calculated with Quantity One Software (BioRad) via the normalization to GAPDH.
ALP activity assay and alizarin red stain analysis
MC3T3-E1 cells were seeded into 96-well plates for ALP activity assay. Cells were incubated with osteogenic medium containing 0, 10−9, 10−8, or 10−7 M Liraglutide for 3, 7, 14, and 21 days, respectively. The control group was incubated with the α-MEM supplemented with 10% FBS; it was incubated with the same amount of physiological saline. Cells were harvested and lysed with protein lysis buffer (Beyotime Biotechnology), and 30 μL samples were quantified with an ALP Detection Kit (Jiancheng Biotech Institute, Nanjing, China) at a wavelength of 520 nm according to the manufacturer's protocol. ALP activity was normalized to total protein and expressed as IU/mg.
MC3T3-E1 cells were seeded into a six-well plate for mineralization assay. Cells were incubated with osteogenic medium containing 0, 10−9, 10−8, or 10−7 M Liraglutide, respectively. The control group was incubated with the α-MEM supplemented with 10% FBS. After 21 days, cells were washed with PBS and fixed with 70% pre-cooling ethanol for 1 h at room temperature. After PBS washes, cells were stained with 1% alizarin red solution (Sigma) at 37°C for 1 h. Then, the cells were washed with distilled water and captured. Alizarin red was precipitated with 10% cetylpyridinium chloride (Sigma) for 30 min at room temperature. The calcium deposition was determined with a microplate spectrophotometer at the wavelength of 562 nm. All values were normalized to the total protein content.
Cell transfection and inhibition study
MC3T3-E1 cells were seeded into six-well plates for cell transfection. After 24 h, cells were divided into eight groups: SiCon, SiCon+Li, SiSmad2, SiSmad2+Li, SiSmad3, SiSmad3+Li, SiSmad2+SiSmad3, and SiSmad2+SiSmad3+Li. The cells were transfected with siRNA targeted negative control (SiCon), Smad2 (SiSmad2), Smad3 (SiSmad3), or Smad2+Smad3 (SiSmad2+SiSmad3), respectively. After 48 h, SiCon+Li, SiSmad2+Li, SiSmad3+Li, and SiSmad2+SiSmad3+Li groups were treated with osteogenic medium containing 10−7 M Liraglutide. Cells were isolated for qRT-PCR and western blot, ALP activity, and alizarin red stain analysis. The siRNA silencing efficiency was determined by qRT-PCR and western blot. The primers for qRT-PCR were listed as follows: Smad3 forward primer: 5′-accacccagaatatcaaca-3′, reverse primer: 5′-actcgcacaagtctacgg-3′; Smad2 forward primer: 5′-aacgggacaattagatgag-3′, reverse primer: 5′-ccacagtcggcagtagat-3′. The antibodies for western blot were rabbit anti-Smad2, anti-Smad3, anti-GAPDH, and anti-RUNX2 antibodies.
MC3T3-E1 cells were seeded into six-well plates and divided into six groups: control, Li, DKK-1, DKK-1+Li, LY294002, and LY294002+Li. The Li group was administered with osteogenic medium containing 10−7 M Liraglutide. The DKK-1 group was treated with DKK-1 at the final concentration of 0.2 μg/mL. The DKK-1+Li group was pretreated with DKK-1 (0.2 μg/mL) for 3 h and then cultured with 10−7 M Liraglutide. LY294002 cells were incubated with LY294002 at the final concentration of 10 μM. The DKK-1+Li group was pretreated with LY294002 (10 μM) for 1.5 h and administered with osteogenic medium containing 10−7 M Liraglutide. The control group was given the same amount of vehicle. After 3 days, cells were collected for qRT-PCR and western blot. The antibodies for western blot were rabbit anti-AKT (1:1000), anti-p-AKT (1:500), anti-β-catenin (1:500), anti-Smad2, anti-Smad3, anti-p-Smad2, and anti-p-Smad3 antibodies (Abcam).
Statistical analysis
All experiments were independently repeated three times. Data were presented as the mean ± standard deviation and analyzed by using SPSS software version 18.0 (SPSS, Inc.). Statistical analysis was performed by using Student's t-test and one-way ANOVA followed by post hoc methods. p < 0.05 was considered statistically significant.
Results
Liraglutide has no effect on MC3T3-E1 cell proliferation
To explore the effect of Liraglutide on cell proliferation, we performed CCK8 assay and cell cycle testing. In Figure 1A, no differences about cell viability were observed among groups treated with 0, 10−9, 10−8, or 10−7 M Liraglutide for 24 h (p > 0.05). Further, different incubation time (24, 48, and 72 h) had no effect on cell viability (p > 0.05). Then, we further investigated the effect of different concentrations of Liraglutide (0, 10−9, 10−8, or 10−7 M) on cell cycle in Figure 2B. After 72 h of incubation, the percentage of cell numbers in G1, S, and G2/M from four groups (0, 10−9, 10−8, or 10−7 M Liraglutide) displayed no difference (p > 0.05). These results showed that Liraglutide (0, 10−9, 10−8, or 10−7 M) had no effect on MC3T3-E1 cell proliferation after 24, 48, and 72 h of incubation.

Liraglutide has no effect on cell proliferation of MC3T3-E1 cells.

Liraglutide stimulates the expression of GLP-1R in MC3T3-E1 cells.
Liraglutide promotes GLP-1R expression in a dosage- and time-dependent manner
Immunocytochemistry was employed to explore the presence of GLP-1R in MC3T3-E1 cells. Figure 2A showed that GLP-1R was localized in the whole cells, and the signals of GLP-1R were enhanced after Liraglutide incubation. To examine the effect of Liraglutide on GLP-1R expression in MC3T3-E1 cells, we incubated MC3T3-E1 cells with different concentrations of Liraglutide (0, 10−9, 10−8, or 10−7 M) for 3, 7, 14, or 21 days, respectively. In Figure 2B, the mRNA levels of GLP-1R were improved with an increase of Liraglutide concentration (0, 10−9, 10−8, or 10−7 M) under the same incubation time. What is more, similar trends were observed on the protein levels of GLP-1R in Figure 2C and D. Based on the earlier results, 10−7 M Liraglutide was selected as the optimal concentration for further study. As shown in Figure 2E and F, the mRNA and protein levels of GLP-1R were the highest on day 14 at the concentration of 10−7 M Liraglutide. Thus, we concluded that GLP-1R was present in MC3T3-E1 and Liraglutide promoted GLP-1R expression in a dosage- and time-dependent manner in MC3T3-E1 cells.
Liraglutide induces the osteogenic differentiation of MC3T3-E1
To explore the effect of Liraglutide on the osteogenic differentiation of MC3T3-E1, we performed the ALP activity and alizarin red stain analysis, and we detected the expression of osteogenic markers RUNX2 and OCN. In Figure 3A, compared with the control group, the addition of osteogenic medium (0 M Liraglutide) enhanced the ALP activity. Compared with untreated time-matched controls, Liraglutide significantly stimulated the ALP activity on 3, 7, 14, and 21 days of Liraglutide incubation. The effect of Liraglutide on the mineralization of MC3T3-E1 cells was examined by alizarin red staining in Figure 3B and C. After continuous treatment for 21 days, the number of mineralized nodules and the content of calcium deposition were significantly increased in the presence of 0, 10−9, 10−8, or 10−7 M Liraglutide, with a maximal effect at 10−7 M. In Figure 3D and E, the mRNA levels of RUNX2 and OCN were upregulated with an increase of the Liraglutide concentration. Similar trends about the protein levels of RUNX2 and OCN were observed in Figure 3F–H. In Figure 3I, under 10−7 M Liraglutide treatment, the mRNA and protein levels of RUNX2 at 7 days was the highest; however, OCN at 21 days was the highest. Similar trends on the protein levels of RUNX2 and OCN were observed in Figure 3J. So Lireglutide stimulated the osteogenic differentiation of MC3T3-E1.
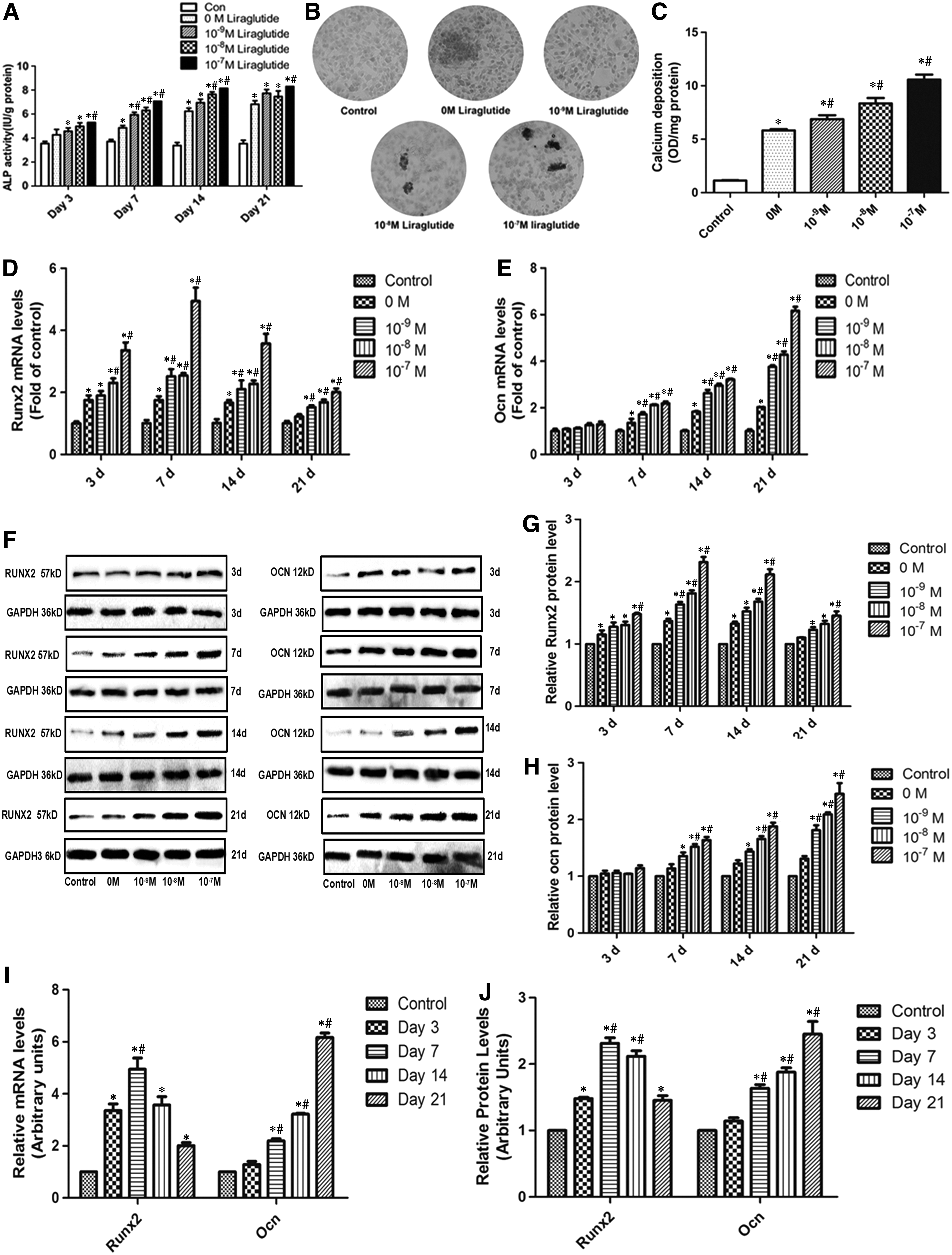
Liraglutide induces the osteogenic differentiation of MC3T3-E1.
Liregluatide promotes the osteogenic differentiation of MC3T3-E1 via regulating Smad2/3
To examine the relationship between Liraglutide and the TGF-β/Smad pathway, we incubated cells with a range concentration of Liraglutide to examine the expression of Smad2/3 in Figure 4. In Figure 4A, compared with control and 0 M groups, 10−9, 10−8, or 10−7 M Liraglutide significantly upregulated the mRNA of Smad2 and Smad3 (p < 0.05). Similarly, Liraglutide promoted the protein levels of Smad2/3 in Figure 4B–D. What is more, the p-Smad2 and p-Smad3 expression was induced, and the p-Smad2/Smad2 and p-Smad3/Smad3 were also upregulated in a dosage-dependent manner. That is to say, Liraglute upregulated the expression of Smad2/3 and the ratio of p-Smad/Smad in MC3T3-E1 cells.
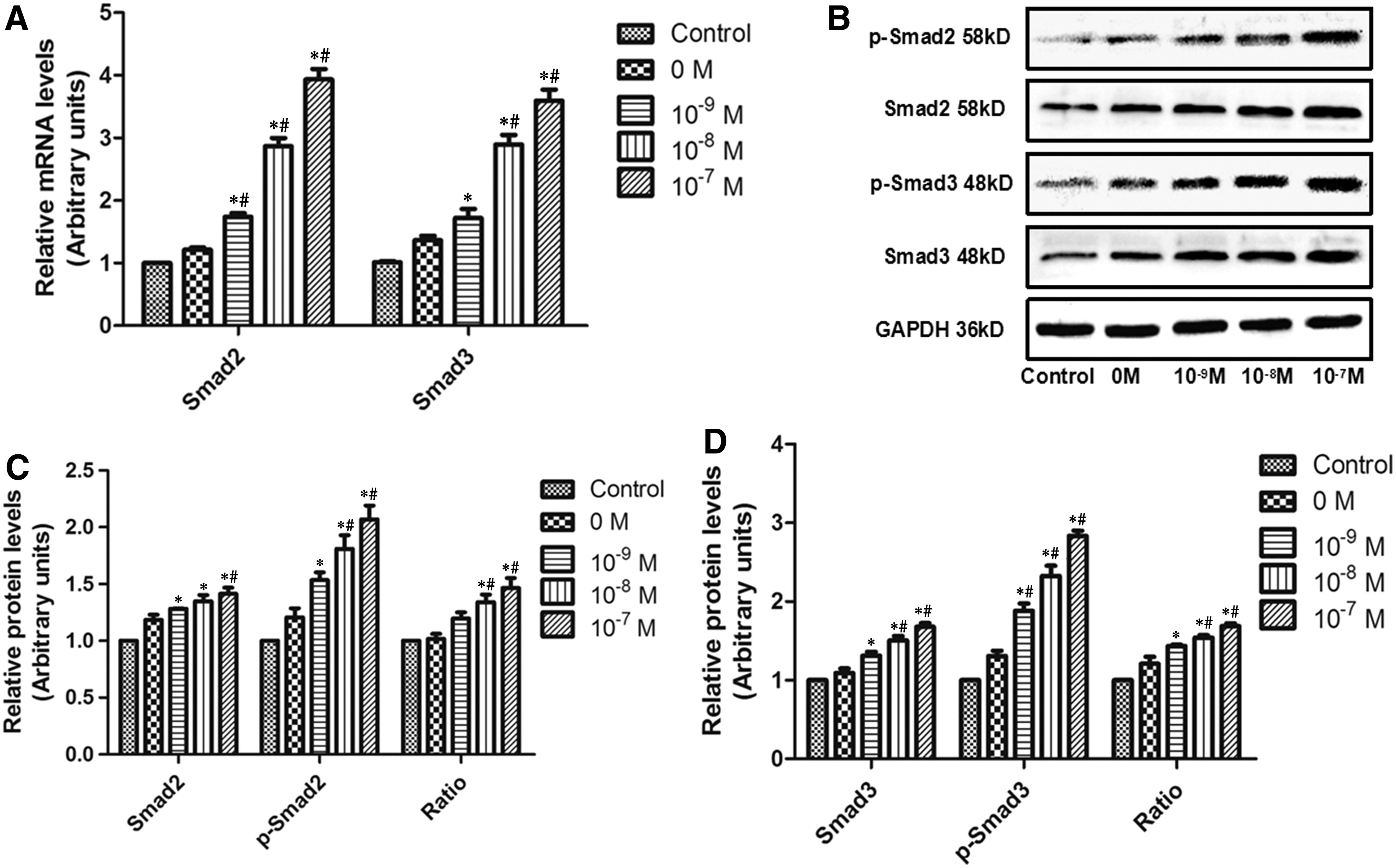
Liraglutide modulates the expression of Smad2/3 in MC3T3-E cells.
To further determine the relationship between Smad2/3 and Liraglutide-induced osteogenic differentiation, we knocked down the levels of Smad2/3. In Figure 5, we found that the transfection of siRNA targeted to Smad2 and Smad3 significantly repressed the mRNA and protein levels of Smad2/3, which provided good evidence for follow-up experiments. In Figure 6A, the addition of Liraglutide significantly enhanced the ALP activity (SiCon+Li > SiCon, SiSmad2+Li > SiSmad2, SiSmad3+Li > SiSmad3, SiSmad2+SiSmad3+Li > SiSmad2+SiSmad3). However, compared with SiCon+Li, the ALP activity in SiSmad2+Li, SiSmad3+Li, and SiSmad2+SiSmad3+Li was obviously blocked, with the maximum effect in the SiSmad2+SiSmad3+Li group. Similarly, SiSmad2, SiSmad3, or SiSmad2+SiSmad3 transfection suppressed Liraglutide-induced upregulation on RUNX2 mRNA and protein levels (Fig. 6B–D). After continuous treatment for 21 days, compared with the SiCon+Li group, the number of mineralized nodules and the content of calcium deposition after siRNA transfection under the condition of Liraglutide were significantly decreased (p < 0.05, Fig. 6E, F). Taken together, the silencing of Smad2/3 blocked the osteogenic differentiation induced by Liraglutide in MC3T3-E1.
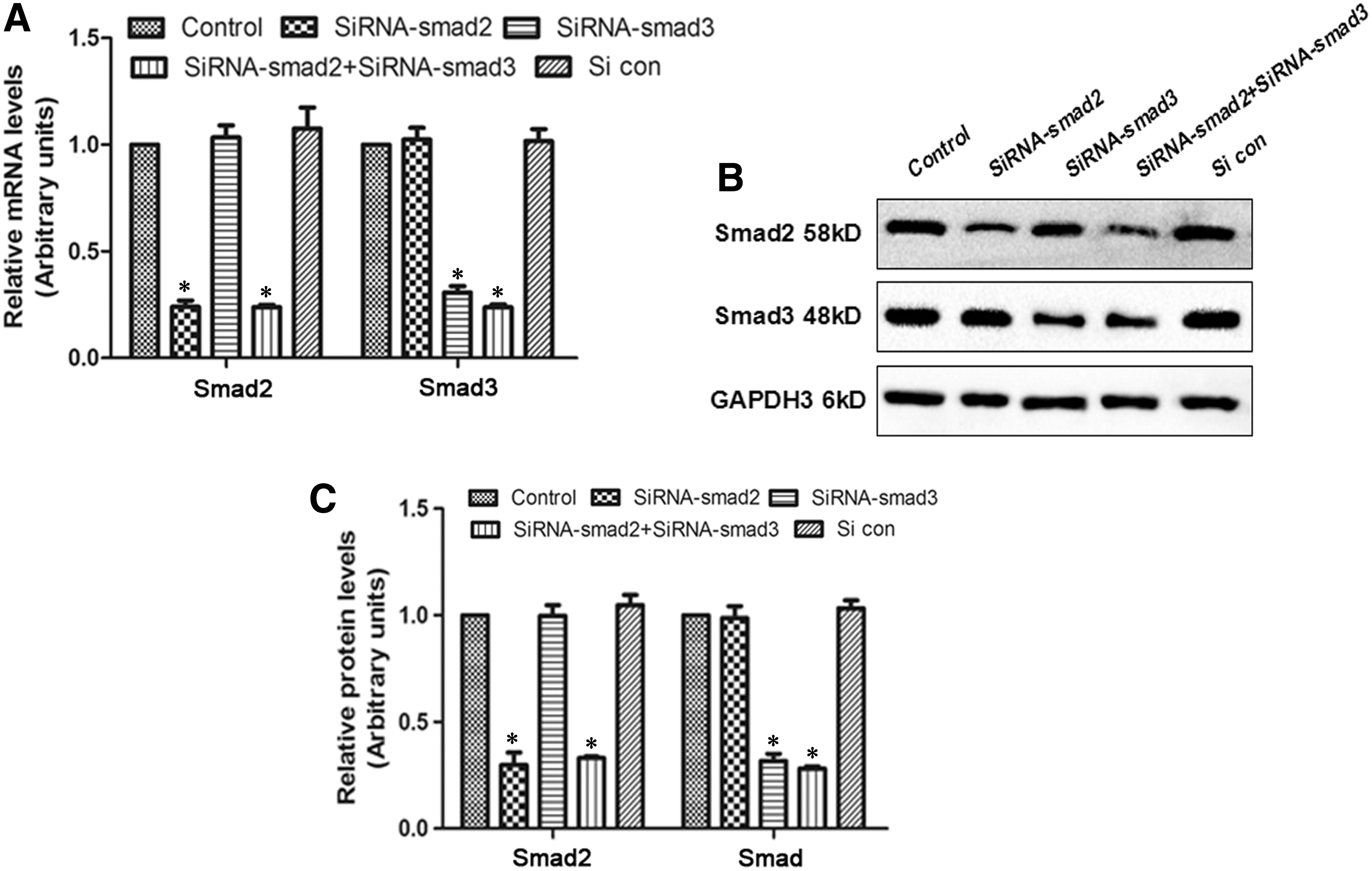
SiRNA transfection successfully blocked the expression of Smad2/3.
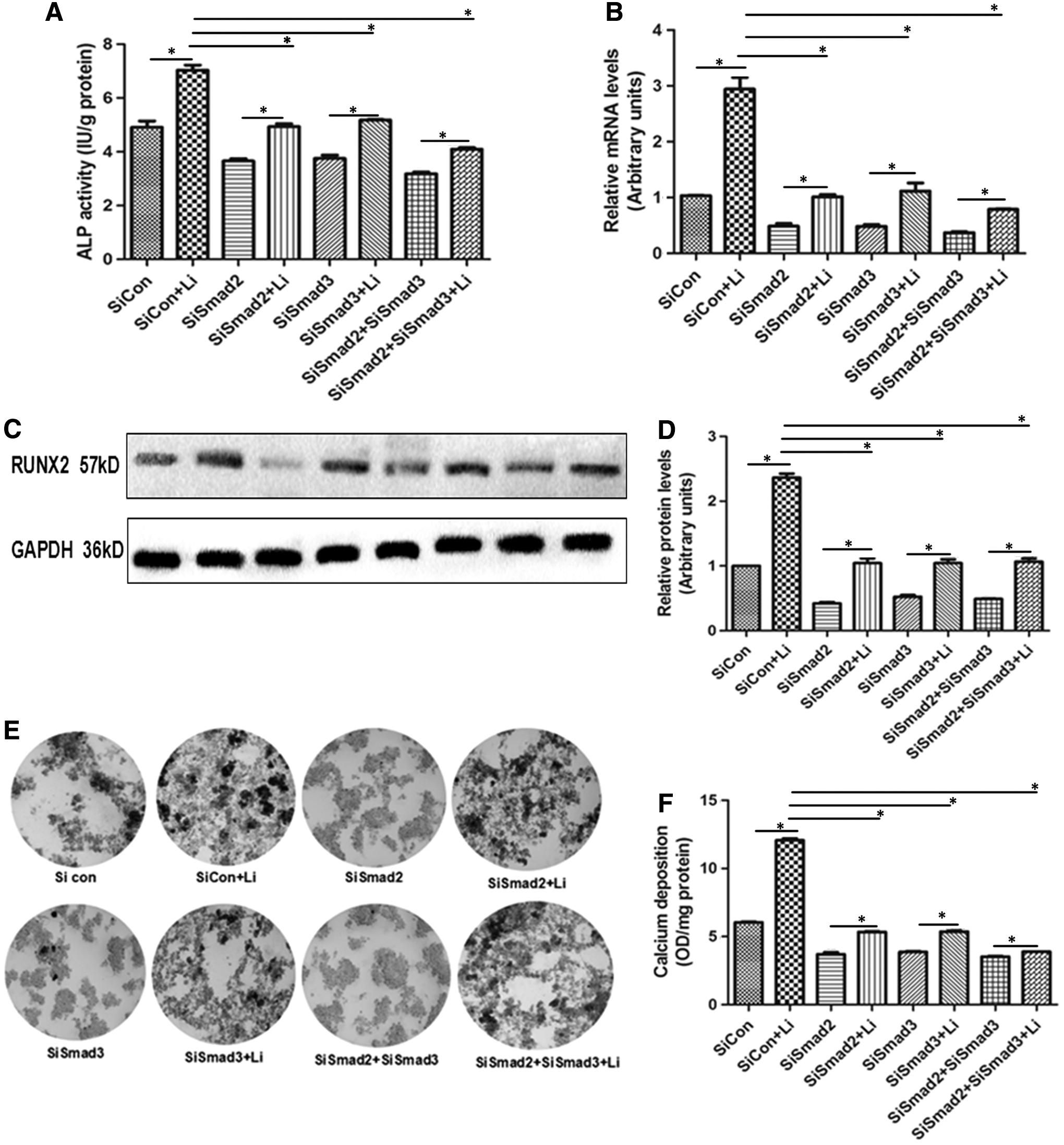
Liraglutide induces the osteogenic differentiation of MC3T3-E1 via regulating Smad2/3.
Liraglutide modulates the expression of Smad2/3 via Wnt/β-catenin and PI3K/AKT pathways
To explore the mechanism by which Liraglutide modulates the expression of Smad2/3 in MC3T3-E1 cells, we employed the PI3K and Wnt inhibitors. In Figure 7A and 7B, the application of Liraglutide improved the expression of AKT and the phosphorylation of AKT (p-AKT) (Li > control, LY+Li > LY). However, the pretreatment of LY significantly reduced the enhancement of AKT and p-AKT induced by Liraglutide (LY+Li < Li). In Figure 7C and D, the relative protein expression of β-catenin in the Li group was higher than that in the control group. However, compared with the Li group, the addition of DKK-1 or LY obviously repressed the upregulation of β-catenin caused by Liraglutide (p < 0.05). Similar trends were observed on the levels of Smad2, p-Smad2, p-Smad2/Smad2, smad3, p-Smad3, and p-Smad3/Smad3 (Fig. 7C, F). In Figure 7C, we further examined the effect of inhibitors on the mRNA of Smad2/3 in Figure 7G. The application of DKK-1 or LY under the incubation of Liraglutide significantly abolished the upregulation of Smad2/3 mRNA induced by Liraglutide. These data indicated that Liraglutide regulated the expression of Smad2/3 through the activation of Wnt/β-catenin and PI3K/AKT pathways.
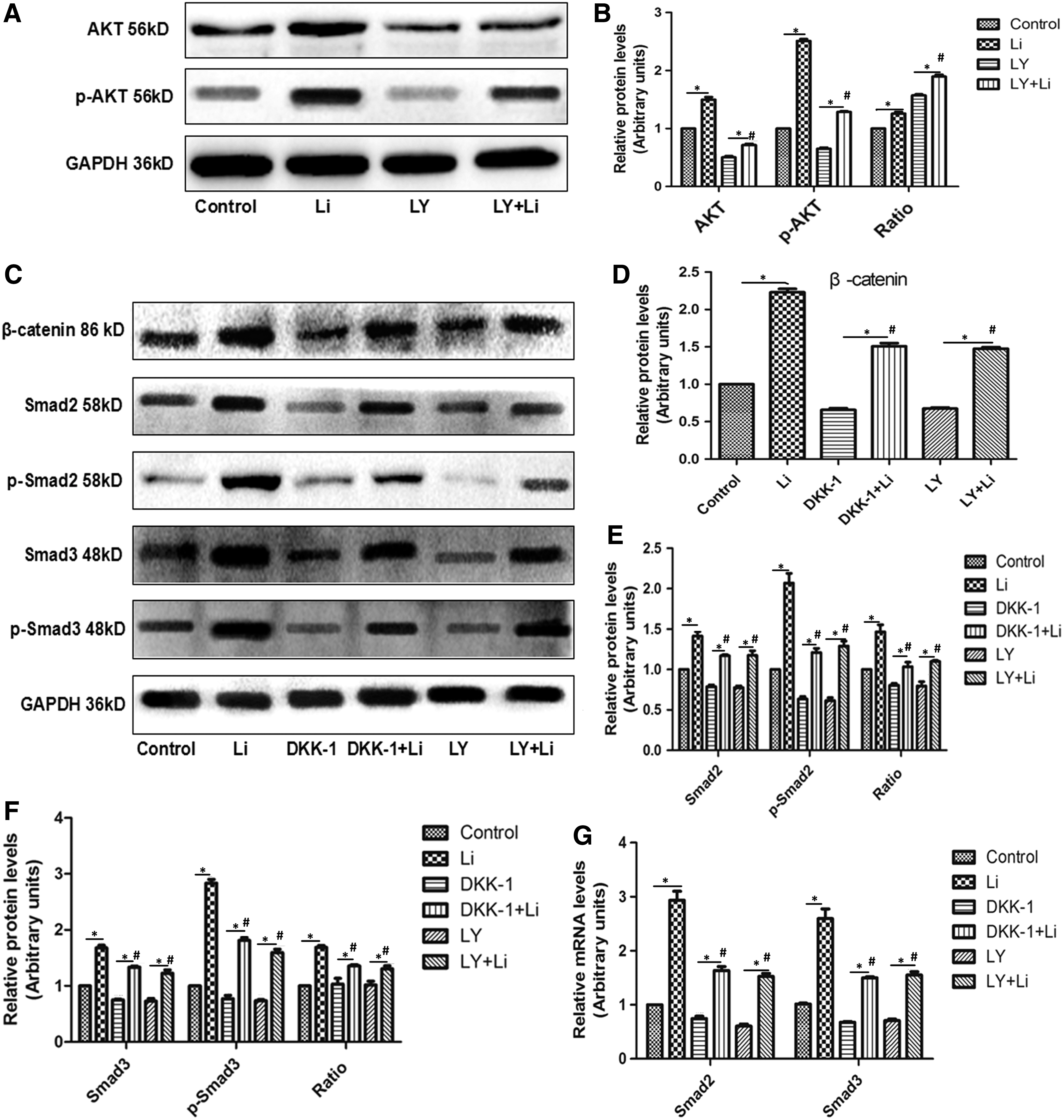
Liraglutide modulates the expression of Smad2/3 via Wnt/β-catenin and PI3K/AKT pathways.
Discussion
Overactive bone resorption or dysfunction of bone formation causes the imbalance between bone resorption and formation and results in the structural integrity and strength of bone (Sanghani-Kerai et al., 2018). Bone metabolism disorder affects a large proportion of the elderly population and is associated with high morbidity and economic burden. GLP-1 and GLP-1RA play a positive important role in facilitating bone formation and suppressing bone resorption (Nauck and Meier, 2018). Recently, several studies indicated that the drug Liraglutide exerts osteogenic and anti-resorptive effects (Ramsey and Isales, 2017). Our current study investigated the effects of Liraglutide on the osteogenic differentiation and explored the underlying mechanism by which Liraglutide promoted the osteogenic differentiation in MC3T3-E1 cells. These findings revealed a new mechanism by which Liraglutide promoted osteogenic differentiation and provided theory evidence to maintain normal bone metabolism during diabetes therapy.
Previous studies showed that GLP-1 exerts its biological activities via binding to the GLP-1R (Kazafeos, 2011). In our research, GLP-1R was confirmed to be present in MC3T3-E1 cells by immunocytochemistry, qRT-PCR, and western blot. These results were in agreement with most previous studies (Aoyama et al., 2014; Wu et al., 2018), but they were contrary to the data reported by Nuche-Berenguer and colleagues (Nuche-Berenguer et al., 2010). The different results might be caused by differences in primers (Wu et al., 2017); however, the exact reasons need more evidence. Wu and colleagues showed that GLP-1R was localized in the surface of MC3T3-E1 cells, but we detected that the signals of GLP-1R were in the whole cells. Further investigations need to be performed in the follow-up study. In addition, the application of Liraglutide significantly improved the mRNA and protein levels of GLP-1R in MC3T3-E1 cells in a dosage- and time-dependent manner. These data provided new evidence about the close relationship between GLP-1R and Liraglutide.
Osteogenic markers such as ALP activity, mineralized nodule, and the expression of RUNX2 and OCN were examined to evaluate the ability of MC3T3-E1 differentiation. The function of Liraglutide on cell differentiation remains elusive. Some reports indicated that Liraglutide facilitates the osteogenic differentiation in MC3T3-E1 cells (Sun et al., 2015; Wu et al., 2017); however, some scholars found that Liraglutide blocks the osteogenic differentiation (Hu et al., 2016). In our study, intracellular ALP activity and the number of mineralized nodules were significantly increased after Liraglutide incubation in a dosage-dependent manner. The mRNA and protein levels of RUNX2 and OCN were also upregulated by Liraglutide. The highest level of RUNX2 was at 7 days of Liraglutide incubation; however, the highest level of OCN appeared at 21 days. The process of osteoblast differentiation consists of three steps: cell proliferation, extracellular matrix formation and maturation, and bone matrix mineralization. During bone matrix maturation, ALP, an early-stage phenotypic marker in the osteoblastic differentiation process (Yu et al., 2017a), increases. The role of RUNX2 is to promote early immature differentiation of osteoblasts, and RUNX2 is the earliest and most specific marker of bone formation (Gu et al., 2018). During bone matrix mineralization, OCN expression increases and the formation of bone nodules indicates bone matrix mineralization (Blair et al., 2017). These data revealed that Liraglutide significantly promoted MC3T3-E1 cells differentiation in different stages, which was in agreement with the first opinion.
The TGF-β/Smad signaling plays a direct and significant role in regulating osteoblast differentiation (Yu et al., 2017b). Smad proteins (Smad2/3/4) are the major transcription factors of TGF-β1 signaling and are involved in TGF-β-related cell responses and pathophysiological functions (Budi et al., 2017). A previous study revealed that Smad2/3 played an important role in the regulation of bone formation (Heo et al., 2018), and the expression of Smad2/3 and p-Smad2/3 was elevated during cementoblast differentiation and mineralization (Li et al., 2015). In our study, we found that Liraglutide significantly upregulated the expression of Smad2/3, p-Smad2/3 and the ratio of p-Smad2/Smad2 and p-Smad3/Smad3, and the silencing of Smad2/3 obviously blocked the osteogenic differentiation induced by Liraglutide. That is to say, Smad2/3 is essential for Liraglutide-induced osteoblastic differentiation. In combination with a previous study, we concluded that Liraglutide facilitated the osteoblastic differentiation via regulating the expression of Smad2/3, which provided new insights for exploring the mechanism of Liraglutide functioning on osteoblastic differentiation.
It is reported that Liraglutide facilitates osteogenic differentiation via the PI3K/AKT pathway (Wu et al., 2017). In our study, we found that the application of Liraglutide promoted the levels of AKT and p-AKT, and the pretreatment of PI3K inhibitor LY294002 suppressed the upregulation of AKT and p-AKT. These results suggested that LY294002 successfully blocked the PI3K/AKT pathway. In PI3K class II-α knock-out mice, the TGF-β-induced Smad2/3 signaling was blocked (Aki et al., 2015). The application of LY294002 extended the duration of Smad2/3 activation and repressed the phosphorylation of Smad2/3 (Yu et al., 2015). In our study, LY294002 also repressed the levels of Smad2/3 and p-Smad2/3 under Liraglutide incubation. Thus, Liraglutide modulated the activation of Smad2/3 via the PI3K/AKT pathway to be involved in osteoblast-mediated bone formation.
Many studies revealed that the Wnt/β-catenin signaling pathways are involved in every bone metabolism process (Yang et al., 2011). In our study, the Wnt inhibitor DKK-1 suppressed the mRNA and protein levels of β-catenin, suggesting that DKK-1 successfully blocked the Wnt/β-catenin pathway. During myofibroblast differentiation, Wnt3α modulated the promoter element binding to Smad2/3 (Yang et al., 2011). Axin and GSK3β in the Wnt/β-catenin pathway stabilized Smad3 protein to modulate the TGF-β pathway (Guo et al., 2008). Smad3 induces the activation of Wnt/β-catenin signaling in mouse osteoblastic cells and promotes the osteoblast differentiation (Liu et al., 2016). These findings revealed the close relationship between Wnt/β-catenin and TGF-β/Smad pathways. In our study, the addition of DKK-1 repressed the activation of Smad2/3 induced by Liraglutide. That is to say, Liraglutide modulated the expression of Smad2/3 via the Wnt/β-catenin pathway for osteoblastic differentiation. In our study, we also found that LY294002 blocked the upregulation of β-catenin induced by Liraglutide, suggesting the crosstalk between Wnt/β-catenin and PI3K/AKT pathways.
Conclusions
We concluded that Liraglutide upregulated the levels of Smad2/3 and p-Smad2/3 via PI3K/AKT and Wnt/β-catenin pathways, in turn promoting osteoblastic differentiation in MC3T3-E1 cells. These findings revealed new mechanisms by which Liraglutide induced osteoblastic differentiation, and they provided theory evidence to maintain normal bone metabolism during diabetes therapy.
Footnotes
Disclosure Statement
No competing financial interests exist.