
Abstract
The globally distributed American cockroach (Periplaneta americana) is considered a pest, but it has been widely used in traditional Chinese medicine. In the past, the American cockroach's genome and transcriptomes were sequenced, but the differential expression transcripts between developmental stages were unavailable. We performed the de novo assembly and analysis of American cockroach transcriptomes from four developmental stages. Approximately 200 million high-quality paired-end reads were generated by using Illumina Hiseq 2000 sequencer. The assembly produced 291,250 transcripts with an average length of 714 bp. In addition, 38,052 microsatellites and 11,060,020 transposable elements were identified. Based on sequence homology, 53,262 transcripts were annotated. After calculating the expression levels of all the transcripts, we found that 13 transcripts were highly expressed in all the samples and at least two, p10 and actin-related protein 1, played important roles during development. A total of 7954 differentially expressed transcripts (DETs) were identified. The adult had the largest number of DETs when compared to other samples (4818), while the 3rd and 8th larva had the least number of DETs (1332). We performed gene enrichment analysis with the DETs, and some interesting results were detected in the different groups. For example, chitin is the major component of the insect exoskeleton, and the chitin-related genes in larvae and new molted samples had higher expression levels than in adults. In addition, the enrichment analysis detected many chitin-related pathways. Our study performed the first large-scale comparative transcriptomics between the developmental stages of American cockroach, which could provide useful gene expression data for future studies.
Introduction
The American cockroach (Periplaneta americana) or simply the “cockroach” can carry a variety of pathogens, and its excrement and shedding of the epidermis are the main sources of indoor allergens (Chen et al., 2015; Salama, 2015; Li et al., 2017). Although the American cockroach is considered a pest and unhygienic, it had been used as a model organism in many fields of science, such as neurobiology (Stankiewicz et al., 2012) and cardiophysiology (Sláma et al., 2006). Furthermore, it has been employed in studies on blood clotting mechanisms (Haine et al., 2007), the discovery of allergenic proteins (Ahmed et al., 2010), the effects of adipokinetic hormones (Wicher et al., 2006), and sexually dimorphic glomeruli and related interneurons (Nishino et al., 2010). The cockroach has also been widely used in traditional Chinese medicine. For example, Kanfuxin liquid was developed using the American cockroach and it is reported to aid in wound healing, immune regulation, treatment of digestive tract ulcers, and tumor adjuvant therapy (Li and Xiao, 2006; Qi and Wu, 2017). However, the effective medicinal ingredients and pharmacological mechanisms of these medical products, including Kanfuxin, are still unclear.
The American cockroach is characterized by incomplete metamorphosis. Nymphal development undergoes several stages, including the early 3rd stage and later 8th stage. Following the 8th larval stage, the newly molted larva is very sensitive to the external environment before finally progressing onto adulthood. The complete genome (Jin et al., 2018; Li et al., 2018) and different transcriptomes of the American cockroach have been sequenced before (Chen et al., 2015; Kim et al., 2016; Zhang et al., 2016), but the differences in gene expression between individuals from different developmental stages are poorly known. Therefore, we aimed to sequence and describe (1) the transcriptome profiles of American cockroaches from four developmental stages (i.e., 3rd larva, 8th larva, newly molted larva, and adult) and (2) the differences between the gene expression of these four developmental stages.
Materials and Methods
Samples
Samples of the American cockroach were provided by the Good-Doctor company (Sichuan, China) in April 2014. Due to similar body size and therefore indistinguishable ages of the 3rd and 4th larva, these were combined. Likewise, the 7th and 8th larva were pooled as well. Consequently, the four developmental stages were 3rd–4th larva (henceforth 3rd larva), 7th–8th larva (8th larva), newly molted larva, and the adult (male and female). Each group had two male and two female individuals, except for the 3rd larval stage. We could not distinguish the gender of the 3rd larva and therefore, we randomly chose four individuals.
RNA extraction and transcriptome sequencing
The total RNA was extracted from the body (without wings, feet, and gut) with Trizol reagent (Life Technologies) in accordance with the manufacturer's instructions. Total RNA was measured using a Nanodrop spectrophotometer (Thermo Scientific) and the quality was assessed with the RNA 6000 Nano assay kit (Agilent) and Bioanalyser2100 (Agilent). A pooled RNA sample of each developmental stage was used for cDNA library preparation. Library construction was completed by Novogene (China), and libraries were sequenced by Illumina HiSeq 2000 platform at Novogene (China).
Sequence preprocessing and transcripts assembly
The 100 bp paired-end (PE) short reads were obtained after sequencing. We filtered out low-quality (70% bases quality higher than Q20) and adaptor-polluted reads. The remaining reads were considered clean reads, which were used for following analyses. The clean reads were combined and de novo assembled with Trinity using the following parameters:—seqType fq—JM 100G–CPU 16, while the remaining were analyzed using default parameters (Grabherr et al., 2011; Haas et al., 2013).
Transcripts annotation
The assembled transcripts were searched against the NCBI nonredundant (nr) protein database, the Swiss-Prot database, and euKaryotic Orthologous Groups (KOGs) with E-value cutoff of 1E-5. BLAST2GO (v2.5) (Conesa et al., 2005) using BLASTX. The resultant transcripts were searched against the Gene Ontology (GO) database. Kyoto Encyclopedia of Genes and Genomes (KEGG) database annotation was performed using the single-directional best-hit (SBH) method in KEGG automatic annotation server (KAAS). KEGG MapperS * was used to reconstruct pathways.
Prediction of open reading frames and ncRNAs
TransDecoder was employed to identify the ORFs (open reading frames) of transcripts with default parameters. Transcripts without ORFs and annotation were searched against the Rfam database using RFAM 11.0 (Griffiths-Jones et al., 2005; Burge et al., 2012) to predict noncoding RNAs (ncRNAs).
Identification of simple sequence repeat and transposable elements
Microsatellite Search and Building Database (MSDB) (Du et al., 2012) was employed to identify SSRs (simple sequence repeats) in the assembled transcriptome for identification of perfect mononucleotide, dinucleotide, trinucleotide, tetranucleotide, pentanucleotide, and hexanucleotide motifs with a minimum of 12, 7, 5, 4, 4, and 4 repeats, respectively. RepeatMasker (v4.0.5) was applied for the identification of DNA-level transposable elements (TEs) using Repbase library with the following parameters: -engine ncbi -nolow -no_is -norna. The protein-level TEs were identified by RepeatProteinMask with the following parameters: -noLowSimple -p-value <0.0001.
Differential expressed transcripts
Expression abundance of transcripts was analyzed using the RSEM (RNA-Seq by Expectation-Maximization), (Li and Dewey, 2011) method for each sample. Furthermore, we used the TMM (trimmed mean of M-values) method to normalize the expression value of each transcript. We then used EdgeR to identify differentially expressed transcripts (DETs). Transcripts with fold change ≥2 and false discovery rate <0.01 were thought to be differentially expressed (Haas et al., 2013). GO enrichment was performed by Gostat (Beißbarth and Speed, 2004), and KEGG enrichment utilized the same method as described by Chen et al. (2010).
Results
Sequencing and assembly
We constructed five cDNA libraries derived from the body (excluding wings, feet, and gut) of American cockroaches at four developmental stages. In total, 217,776,763 PE reads with lengths of 100 bp were generated. After quality control, 200,616,399 PE reads were obtained, which were further assembled into 291,250 contigs with an average length of 714.3 bp (Table 1). All the short reads were deposited in the NCBI SRA database under accession number SRR5286150-SRR5286154.
Summary of Transcripts Assembly and Annotated Transcripts by Protein Database
KEGG, Kyoto Encyclopedia of Genes and Genomes; NR, Non-Redundant Protein Sequence Database.
Functional annotation of transcripts
There were 53,262 transcripts obtained from the annotated samples based on sequence homology (i.e., using Non-Redundant Protein Sequence Database (NR), Swiss-Prot, GO, KOGs, and KEGG databases). Among these, 52,625, 33,168, 15,926, 35,595, and 18,880 transcripts were matched using NR, Swiss-Prot, GO, KOGs, and KEGG databases, respectively (Table 1). The GO annotation showed that the major subcategories were “Cell,” “Cell part,” “binding,” “catalytic,” “cellular process,” and “metabolic process,” respectively (Fig. 1). There were 24,881 transcripts classified into 25 groups using KOGs annotation (Fig. 2). The “General function prediction” represented the most numerous group, followed by “Signal transduction mechanisms” and “Post translational modification, protein turnover, and chaperones.” The smallest number of transcripts was classified as nuclear structures (contains 13 transcripts), cell motility (46 transcripts), and defense mechanisms (159 transcripts). Signal transduction, infectious diseases, and cancers contained the largest numbers of annotated transcripts from the KEGG database (Fig. 3). Moreover, we found a few important KEGG pathways. For example, there were six transcripts within linoleic acid metabolism pathway (Fig. 4) and six transcripts within alpha-linolenic acid metabolism pathway (Fig. 5).

Functional classification of all the transcripts of American cockroach based on Web Gene Ontology Annotation Plot. The results are classed into three main Gene Ontology categories: cellular component, molecular function, and biological process. The left y-axis indicates the percentage of a specific category of genes in that main category. The right y-axis indicates the actual number of genes in a category. Color images are available online.
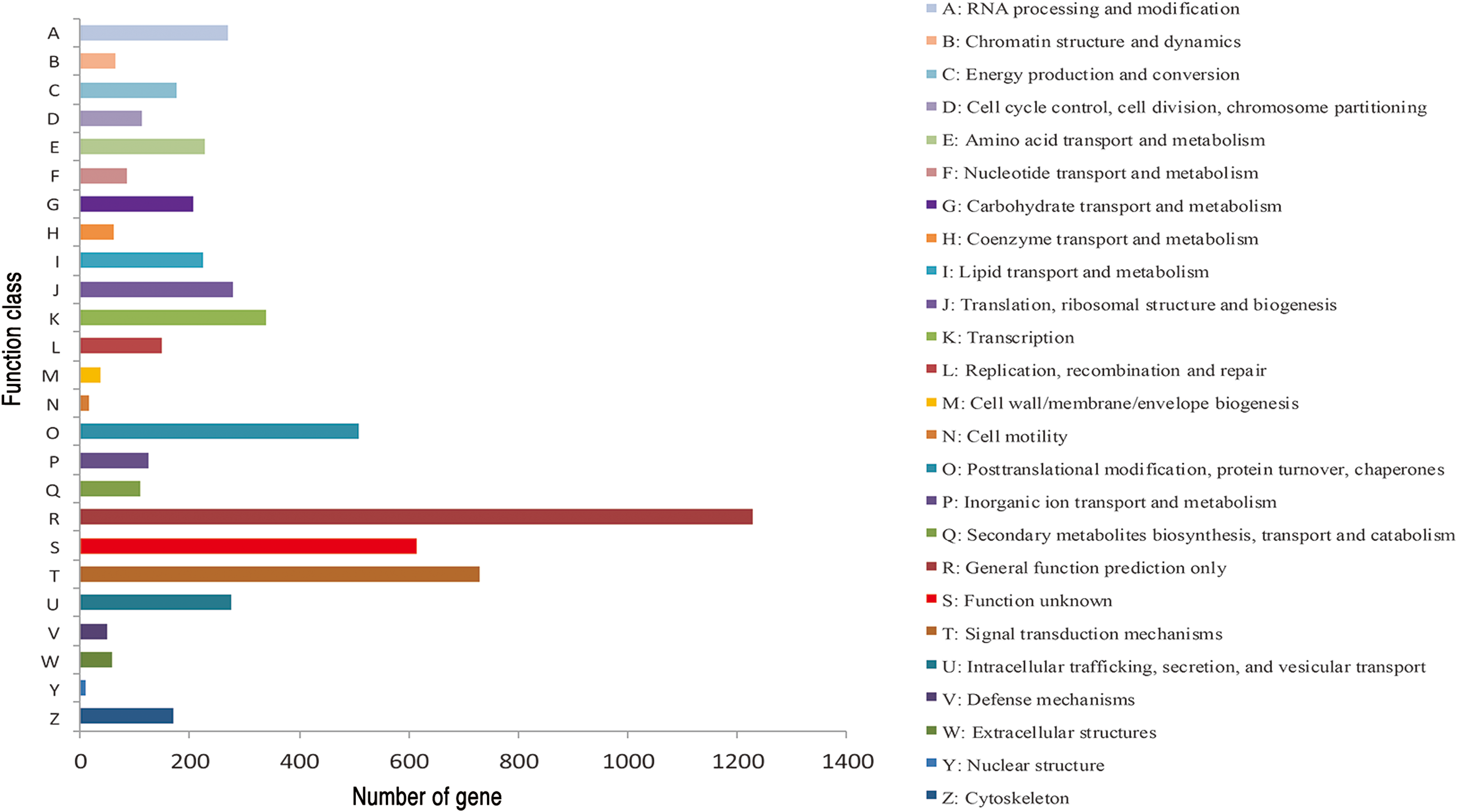
Functional classification of all the transcripts of American cockroach based on euKaryotic Orthologous Groups. In total, 24,881 transcripts were classified into 25 classes. Color images are available online.

Functional classification of all the transcripts of American cockroach based on Kyoto Encyclopedia of Genes and Genomes. Color images are available online.
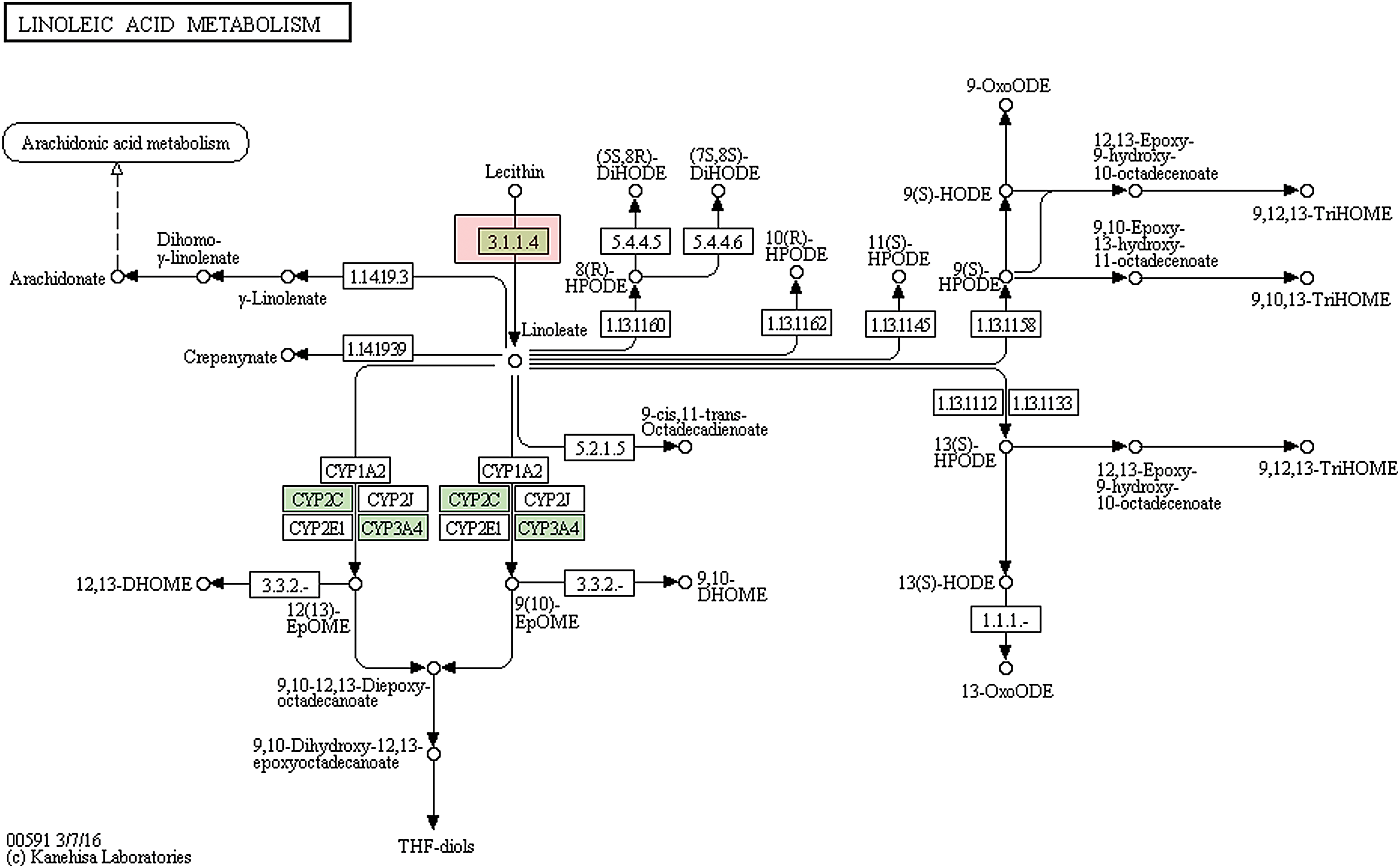
Linoleic acid metabolism pathway. The green box indicates that gene had annotation within the transcripts of American cockroach. There were six transcripts annotated within the linoleic acid metabolism pathway. The PLA2G (EC:3.1.1.4) were upregulated in newly molted larva when compared with other samples. Color images are available online.

Alpha-linolenic acid metabolism pathway. The green box indicates that gene had annotation within the transcripts of American cockroach. There were six transcripts annotated within the alpha-Linolenic acid metabolism pathway. The ACX and PLA2G (EC:3.1.1.4) were downregulated in the adult group when compared with other samples (3rd larva, 8th larva, and newly molted larva). Color images are available online.
Prediction of ORFs and ncRNA
A total of 237,988 transcripts were without annotations and among them, we found 7298 ORFs (Supplementary Data S1). We also predicted 85 ncRNAs from the remaining 230,690 nonannotated transcripts, with 60 of them being tRNAs.
Identification of SSRs and TEs
In total, we identified 38,052 SSRs within found transcripts, with the total length of 617,673 bp and an average length of an SSR being 16.23 bp. The frequency and density of SSRs were 193.83 loci/Mb and 2983.93 bp/Mb, respectively. Mononucleotide SSRs were the most abundant category, followed by dinucleotide > tri nucleotide > tetranucleotide > pentanucleotide > hexanucleotide SSRs (Table 2). TE analysis detected 11,060,020 TEs in transcripts, and among them, DNA transposon was the most numerous group (7,259,649), followed by long interspersed nuclear elements (2,910,446), long terminal repeats (857,213), short interspersed nuclear elements (32,433), and other types (279). Subsequent analysis of transcript expression showed that a total of 15,522 transcripts containing TEs were expressed, with fragments per kilobase of transcript per million fragments mapped greater than one in at least one sample.
Summary of Simple Sequence Repeat Unit
DETs and functional distribution
We calculated the expression levels of all transcripts in each sample and collected the 46 transcripts that had the highest expression level in each sample. Of these 46, we obtained transcripts that were shared in all the samples. We found that 13 transcripts were highly expressed across all the samples and a few among them were quite important, such as p10, actin-related protein 1, and elongation factor 1-alpha (Table 3).
Commonly Shared Highly Expressed Transcripts in All the Samples
Afterward, DETs between samples were obtained. A total of 7954 significant DETs were detected by EdgeR (Table 4; Fig. 6). DETs with higher expression levels in one sample when compared to another were denoted as “upregulated,” while those with lower expression levels were termed as “downregulated.” In general, the 3rd larva and 8th larva had the least numbers of DETs (1332) and the adult stage had greatest numbers of DETs. There were 4818 DETs and 3448 DETs found between the adult and 3rd larva and adult and 8th larva, respectively. We also found 3533 DETs between the adult group and newly molted larva (Table 4).

Heatmap of all differentially expressed transcripts in all samples. The larva samples are more close to each other than to other samples. Color images are available online.
The Numbers of Differential Expressed Transcripts Between Samples
GO enrichment analysis with DET comparisons across developmental stages used adult samples without the inclusion of gender separately. We combined the female and male adult samples within the adult group because other groups contained both female and male samples. At first, we investigated the 4818 DETs identified between adult and 3rd larva. The upregulated transcripts in adult samples included enrichment in hydrolase activity, response to other organisms, and stress response (Supplementary Table S1). However, the downregulated transcripts in adult samples were mainly enriched in cuticle structural constituents and several extracellular-related categories (Supplementary Table S2). Likewise, we performed GO enrichment with the 3448 DETs between adult and 8th larva. The upregulated transcripts in adult samples were enriched in peroxidase activity, oxidoreductase activity, and hydrolase activity (Supplementary Table S3). Upregulated transcripts in 8th larva were enriched in cuticle structural constituents, nutrient reservoir activity, and extracellular matrix structure constituents (Supplementary Table S4). Furthermore, we compared the 1953 DETs found between 8th larva and newly molted larva. The upregulated transcripts in 8th larva were enriched in cuticle structural constituents, peptidase activity, and cell cycle-related categories (Supplementary Table S5). The downregulated transcripts in 8th larva were also enriched in cuticle structural constituents (Supplementary Table S6). We then performed GO enrichment analysis with the 1675 DETs identified between newly molted larva and 3rd larva (Supplementary Tables S7, S8). The upregulated transcripts in newly molted larva were mainly enriched in cuticle-related categories, such as structural constituent of cuticle, cuticle development, chitin binding, and chitin metabolic process (Supplementary Table S7). The upregulated transcripts in the 3rd larva were mainly enriched in several categories of peptidase activity (Supplementary Table S8). In addition, we compared the 1332 DETs between the 3rd larva and 8th larva (Supplementary Tables S9, S10). The upregulated transcripts in 8th larva were mainly enriched in several microtubule-related categories and protein-related categories (Supplementary Table S9), whereas the upregulated transcripts in 3rd larva were mainly enriched in signaling pathways (Supplementary Table S10). Finally, GO enrichment analysis was performed with the 3533 DETs between adult and newly molted larva (Supplementary Tables S11, S12). The upregulated transcripts in adult were mainly enriched in odorant binding, hydrolase activity, and nutrient reservoir activity (Supplementary Table S11), whereas the upregulated transcripts in newly molted larva were mainly enriched in carbohydrate derivative binding and photosystem (Supplementary Table S12).
Discussion
Our de novo assembly of American cockroach transcriptomes from four developmental stages found 200,616,399 high-quality PE reads and 291,250 transcripts with an average length of 714 bp. We identified 7298 ORFs, and predicted 85 ncRNAs, which can act as a valuable resource for improving the gene annotation of the American cockroach. The functional annotations of the assembled transcriptome found that the general pattern was very similar with the results of the Hemimetabolous German cockroach (Blattella germanica) (Zhou et al., 2014). However, there were many transcripts that were annotated in important KEGG pathways, such as linoleic acid metabolism pathway (Fig. 4) and alpha-linolenic acid metabolism pathway (Fig. 5).
Chen et al. (2015) did not find transcripts associated with “Extracellular structures” in the testis transcriptome, but we found 58 annotated transcripts related to this group (Fig. 2). Chen et al. (2015) only collected the testes from 10 males for sequencing, but we had samples from the four developmental stages of the American cockroach, which was useful for finding more transcripts. Besides, we had more than 200 million reads, but there were only ∼65 million reads in Chen et al. (2015). Therefore, the sampling and sequencing reads probably contributed to the observed differences between our study and Chen et al. (2015).
Our transcriptomes provided gene expression profiles and unique resources to select the genetic makers for population genetics and genetic management (Vasemägi et al., 2005; Chen et al., 2015; Du et al., 2015). For example, Du et al. (2015) identified more than 20,000 transcriptome-derived microsatellites (SSRs) in the giant panda's (Ailuropoda melanoleuca) blood transcriptome. Chen et al. (2015) detected 14,195 potential SSRs in the testis transcriptome of the American cockroach (Chen et al., 2015). In our study, we identified 38,052 SSRs in the transcripts due to the utilization of more samples than Chen et al. (2015), which increased the SSR database for the American cockroach. Besides SSRs, we detected 11,060,020 TEs in transcripts. Some TEs, such as Alu element and CR1 element, have been used as genetic markers to reconstruct the phylogenetic relationships of different species (Guo et al., 2015; Cui et al., 2016). Therefore, the SSR and TE data represent important resources for future studies related to population genetics, diversity, and genetic research on adaptive traits.
We calculated the expression levels of transcripts and detected the DETs between different developmental stages of American cockroaches. We found that 13 transcripts were highly expressed in all the samples and detected three important transcripts: protein p10, actin-related protein 1, and elongation factor 1-alpha (Table 3). Protein p10 had a very high expression level in the regenerating limbs and it is also expressed in the antennae and heads of nymphal and adult cockroaches, which was thought to be associated with regeneration (Kitabayashi et al., 1998; Nomura et al., 2002). Actin-related protein 1 (Arps1) was also highly expressed in the samples. The actin-related proteins (Arps) participate in a diverse array of cellular processes, which is crucial for the cell (Schafer and Schroer, 1999). These highly expressed transcripts in the samples might play important roles in the development of the American cockroach and in maintaining the normal life cycle.
In total, 7954 DETs were identified. The adult group had the greatest number of DETs when compared to other samples (from 3448 to 4818). Meanwhile the 3rd larva and 8th larva had the least number of DETs (1332) (Table 4). Some genes and pathways need to be further studied. For example, chitin is the major component of the insect exoskeleton and cockroaches molt several times during its development. The chitin-related genes in larvae and new molted samples had higher expression levels than adults and the enrichment analysis also detected many chitin-related pathways. Through large-scale transcriptome sequencing, we studied the gene expression between the different developmental stages in the American cockroaches. Our findings can provide useful gene expression data for further research.
Funding Information
This project was supported by the standardization of traditional Chinese medicine in National Administration of Traditional Chinese Medicine (ZYBZH-C-SC-53).
Footnotes
Disclosure Statement
No competing financial interests exist.
Supplementary Material
Supplementary Data S1
Supplementary Table S1
Supplementary Table S2
Supplementary Table S3
Supplementary Table S4
Supplementary Table S5
Supplementary Table S6
Supplementary Table S7
Supplementary Table S8
Supplementary Table S9
Supplementary Table S10
Supplementary Table S11
Supplementary Table S12
References
Supplementary Material
Please find the following supplemental material available below.
For Open Access articles published under a Creative Commons License, all supplemental material carries the same license as the article it is associated with.
For non-Open Access articles published, all supplemental material carries a non-exclusive license, and permission requests for re-use of supplemental material or any part of supplemental material shall be sent directly to the copyright owner as specified in the copyright notice associated with the article.