
Abstract
Ischemia-reperfusion injury is a major reason for acute kidney injury and various kidney diseases. Autophagy plays an important role during renal ischemia-reperfusion injury (RIRI), but it remains controversial whether autophagy contributes to cell survival or ischemia-reperfusion-induced cell death. In the review, we summarized the function of autophagy in the progression of acute ischemic kidney injury, as well as its related molecular mechanisms. While analyzing the opposite roles of autophagy in RIRI, it was concluded that the protective or detrimental function of autophagy was depending on the timing and amount of the activation of cell autophagy. We also summarized the regulatory agents, including active compounds, proteins, or microRNAs (miRNAs), which regulated the cell autophagy during renal acute ischemic kidney injury process. This explained why the opposite conclusion occurred when cell autophagy was studied in the RIRI models from different researchers. Therefore, the article provided a hypothesis to control cell autophagy at the appropriate timing and intensity so as to alleviate renal injury and sustain cell survival of the renal cell.
Introduction
Renal ischemia-reperfusion injury (RIRI)
RIRI is one of the main causes of acute kidney injury (AKI) and acute renal failure (ARF). AKI is one of the most common critical illnesses in clinical practice and has a high mortality rate in clinical practice (Manoeuvrier et al., 2017). Studies have shown that the incidence of AKI in general hospitalized patients, in developed countries, is about 3.2–9.6%; the mortality rate is accounting for 20% of hospitalized patients, and as high as 50% of intensive care unit patients (Samimagham et al., 2011; Yokota et al., 2018). A number of epidemiological surveys have shown that the incidence of AKI in China is increasing year by year, and about 19% of patients eventually need renal replacement therapy (Rodrigues et al., 2013). In addition, AKI is also the most important cause of clinical kidney disease. The molecular indication of AKI includes the increased serum creatinine (Scr) concentration, the highly swollen mitochondria in renal tubular epithelial cells, the reduced mitochondrial cristae with disordered arrangement, even disintegrated, and the newly formed vacuolization in cells (Inal et al., 2014; Hatakeyama et al., 2018). Especially, the acute tubular necrosis is the most serious symptom, which can cause ARF or kidney transplant failure. Therefore, it is particularly important to actively and effectively prevent RIRI (Rocha et al., 2015). However, no specific drugs have been found to prevent and effectively treat RIRI till now.
In recent years, there have been more and more studies on the pathophysiological mechanisms and preventive treatment methods of RIRI. The progression of RIRI involves the variation in organic, cellular, or molecular levels (Ying et al., 2012; Koratala and Kamboj, 2018). Autophagy, as a newly discovered form of programmed cell death, is of great significance for cell survival and cell death after renal ischemia-reperfusion (I/R) (Jia et al., 2018). Therefore, the regulatory mechanism of autophagy in renal cells after RIRI is expected to provide an important intervention target for the prevention and treatment of RIRI (Mei et al., 2016; Melk et al., 2016). In this review, we summarized the possible molecular mechanisms and preventive measures of autophagy in RIRI, and compared the protective or detrimental function of autophagy, which was depending on the timing and amount of activation of cell autophagy. Moreover, we analyzed the methodological procedures and experiment models used in different studies and drew a hypothesis to control the autophagy at the appropriate timing and intensity so as to alleviate renal injury, which provided a reference for more in-depth study of autophagy in the field of kidney disease.
Description of Cell Autophagy
Autophagy is an evolutionary conserved process that is a lysosome-dependent catabolic process to eliminate aging organelles and macromolecular proteins that provide raw materials and nutrients for organelle renewal and cell repair (Deretic and Klionsky, 2018; Jin et al., 2018). It plays a key role to maintain cellular homeostasis and is normally trigged by nutrient deprivation and cell stressors, such as hypoxia, osmotic, and reactive oxygen species (ROS). (Rasmussen et al., 2011). Till now, it has been found that cell autophagy plays an important role on cell survival and cell death during pathologic stress in the kidney, such as ischemia-reperfusion injury (IRI) (Decuypere et al., 2015).
Autophagy is normally divided into three types: macroautophagy, microautophagy, and chaperone-mediated autophagy, depending on the way intracellular substrate is transported to lysosomes (Fig. 1) (Hu et al., 2014; Li et al., 2015). Macroautophagy is mainly responsible for the degradation of long-lived proteins and subcellular organelles (referred as autophagy hereafter). Microautophagy is usually thought to be a direct transport of substrates into lysosomes for degradation, while chaperone-mediated autophagy is a chaperone-mediated transport of the substrates to the lysosomes by a receptor (Ferrucci et al., 2018. Autophagy is mainly characterized by the vesicular sequestration and degradation of cytoplasmic components. In this article, we only discuss the macroautophagy hereafter and the major autophagy cascades are depicted in Figure 1. The cell autophagy is conserved by several main steps, including induction, vesicle nucleation and elongation, autophagosome, and autolysosome formation (Sibirny, 2011; Hurley and Schulman, 2014). Now, more than 40 autophagy-related genes (Atg) are reported in the process of autophagy (Kumar et al., 2014; de Iriarte Rodriguez et al., 2015), such as Atg1, Beclin 1, Atg12, Atg7, Atg8, ATG13, and Atg9 (Kunchithapautham and Rohrer, 2007).
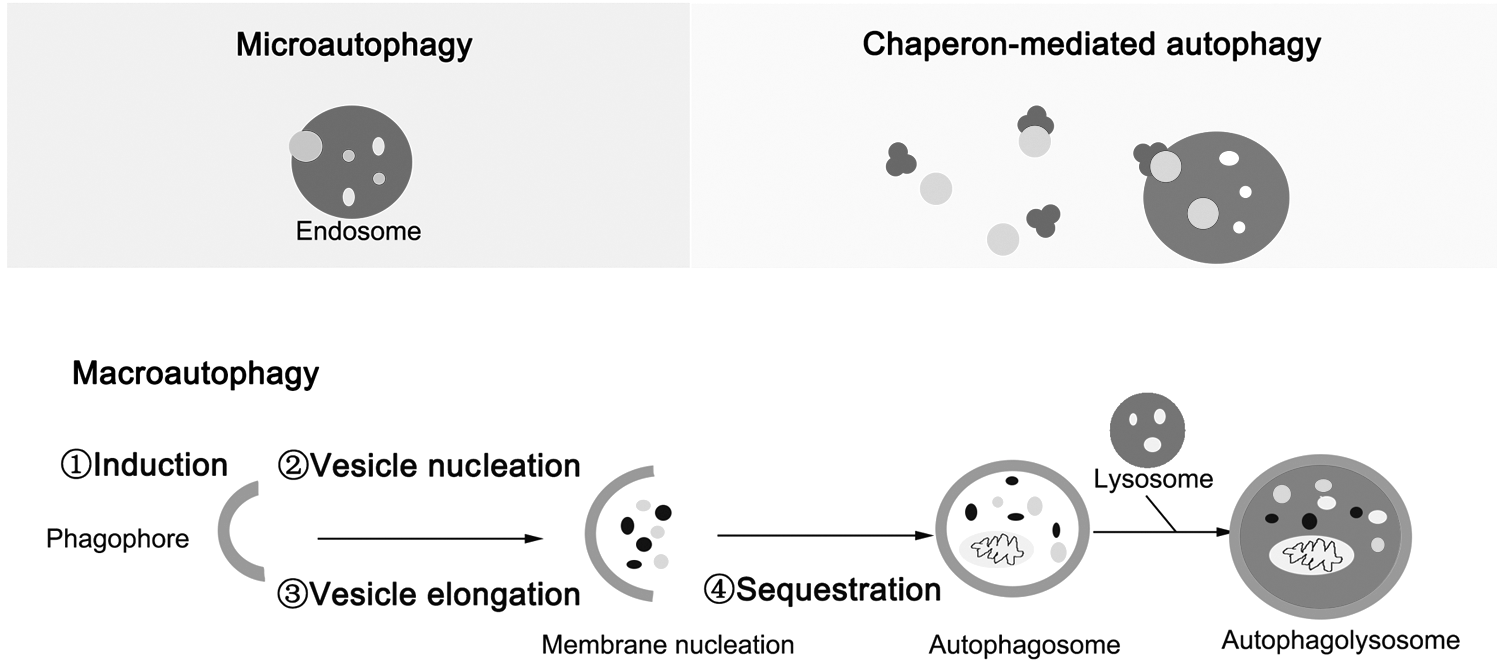
Schematic representation of signaling pathway to cell autophagy. Autophagy is a lysosome-dependent catabolic process to eliminate aging organelles and macromolecular proteins. There are mainly three types of cell autophagy, including microautophagy, chaperon-mediated autophagy, and macroautophagy. In this article, we focus on macroautophagy. The major macroautophagy cascades include several successive steps, such as induction, vesicle nucleation, vesicle elongation, autophagosome formation, fusion, and autolysosome formation. Till now, more than 40 autophagy-related genes (Atg) are reported in the process of autophagy, including Atg1, Beclin 1, Atg12, Atg7, Atg8, ATG13, and Atg9.
Accumulating studies show that there exists crosstalk between cell autophagy and cell apoptosis. In fact, the crosstalk between apoptosis and autophagy during IRI is very complex. Under different time courses of IRI, autophagy has distinct regulatory function on cell survival and death. Sometimes autophagy inhibits apoptosis, while sometimes autophagy promotes apoptosis, involving different signals. For example, some proteins, such as p53, cFLIP, and p62, are traditionally thought to be the key proteins to regulate cell apoptosis, which are also found to regulate cell autophagy now. In addition, the activated PI3K-Akt signaling pathway promotes the activation of cell autophagy and inhibits cell apoptosis, as well by regulating apoptosis-related molecules Bcl-xl, caspase-9, and so on. (Hao et al., 2018). As we know, the tumor suppressor p53 plays an important role in regulating both apoptosis and cell autophagy (Fig. 2). Normally, under physiological conditions, p53 functions as a tetramer and exists at a low expression level in the cells due to its relatively short half-life time (about 20 min), while in the stress state, such as DNA damage, hypoxia, and oncogene activation, the transcriptional activity of p53 is significantly enhanced and the expression level of p53 is highly increased, as well as its prolonged half-life span. Accumulating evidence shows that p53 significantly promotes cell apoptosis by activating the expression of proapoptotic genes, including BAX, apoptotic protease activating factor-1 (APAF-1), p53 upregulated modulator of apoptosis (PUMA), and p53 regulated apoptosis-inducing protein 1 (p53AIP1), and inhibits the expression of antiapoptotic genes BCL-2 and BCL-xL. In addition, p53 also directly promotes apoptosis by binding to BAK on the mitochondrial surface to induce the oligomerization and activation of BAK. However, p53 has a dual effect on autophagy, which can induce cell autophagy and also inhibit autophagy, depending on its subcellular localization. Specifically, nuclear p53 plays a vital role to activate cell autophagy and functions as a transcription factor to transactivate several proautophagic genes, such as AMP-activated protein kinase (AMPK) and DNA-damage regulated autophagy modulator (DRAM). For example, p53 transactivates the β1 and β2 subunit of AMPK; the activated AMPK translocates into cytoplasm and phosphates tuberous sclerosis complex proteins 1 (TSC1), and tuberous sclerosis complex protein 2 (TSC2) to inhibit mammalian target of rapamycin (mTOR), which promotes the activation of cell autophagy. On the other hand, nuclear p53 also induces cell autophagy by transcriptional activation of the lysosomal protein DRAM. Conversely, cytoplasmic p53 inhibits cell autophagy, especially when the nuclear positioning signal is deleted. The cytoplasmic p53 may directly regulate cell autophagy through the endoplasmic reticulum (ER) related pathway, and the regulatory mechanism on cell autophagy is totally different from that of nuclear p53. Interestingly, the inhibition of cell autophagy induced by cytoplasmic p53 is strictly dependent on cell cycle, which mainly occurs at G0/G1 phase, is less found at the S phase, and hardly occurs at the G2/M phase. Moreover, the inhibitory effects of cell autophagy on cytoplasmic p53 may be regulated by microRNAs (miRNAs), such as miR-125b, miR-214, and miR-380-5p (Su et al., 2015; Ferrucci et al., 2018).
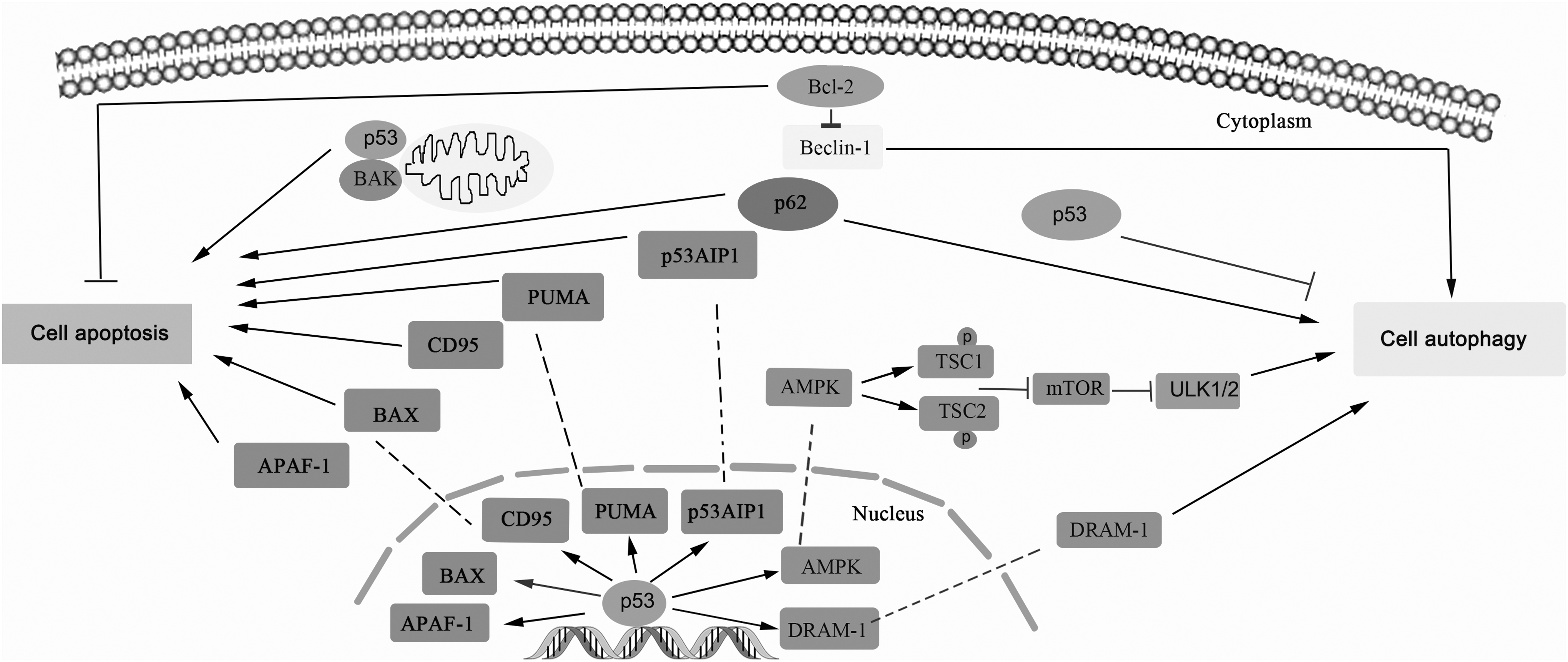
The crosstalk between cell autophagy and apoptosis. Many studies depict a close relationship between cell autophagy and apoptosis. Several proteins are reported to participate in regulating cell autophagy and apoptosis. For instance, p53 plays an important role in regulating both apoptosis and cell autophagy; Beclin-1 suppresses cell apoptosis, while it promotes the process of cell autophagy; p62 promotes both cell autophagy and cell apoptosis during the cell physiological process. AMPK, AMP-activated protein kinase; DRAM, DNA damage-regulated autophagy modulator; mTOR, mammalian target of rapamycin; TSC1, tuberous sclerosis complex proteins 1; TSC2, tuberous sclerosis complex protein 2.
The Autophagy Is a Hot Topic in RIRI Research
In recent years, autophagy has become a new research hotspot in the field of biology and has been studied more and more extensively in the progression of kidney diseases (Lin, 2017). It has been reported that in the pathophysiology of kidney, cell autophagy is closely related to the natural cells of the kidney, such as podocytes, mesangial cells, endothelial cells, and renal tubular epithelial cells, suggesting that autophagy might play an important role in a variety of kidney diseases (Pallet and Anglicheau, 2009). RIRI is a common clinical pathological process, which is the most common cause of ARF. More and more studies have found that autophagy is induced in the RIRI model (Hu et al., 2018; Lim et al., 2019). In this study, we summarized the role of autophagy in different models of RIRI and compared the different studies to discuss the function of autophagy in the process of IRI.
Autophagy activation attenuates RIRI
IRI is a major reason for AKI, which lacks effective clinical therapies till now. Activation of autophagy may provide a protective mechanism for cell survival and a potential strategy for the clinical therapy of AKI, as indicated by renal function, histology, and tubular apoptosis (Fig. 3) (De Rechter et al., 2016; Song et al., 2018). For instance, I/R pretreated with autophagy activator rapamycin alleviated renal injury with improved renal function and inhibited cell apoptosis, while inhibition of autophagy by 3-methyladenine (3-MA) before I/R increased renal injury, with worsened renal function and more tubular apoptosis. Autophagy was detected to be activated during I/R injury and contributed to a protection in RIRI (Zhang et al., 2015). In renal pathophysiology, inhibition of autophagy by chloroquine and 3-MA worsened RIRI (Jiang et al., 2010). Moreover, suppression of autophagic activation by impaired AMPK/ULK1 signaling in proximal tubules worsened type 2 diabetes mellitus-induced RIRI (Muratsubaki et al., 2017). The activation of PINK1-PRKN/PARK2 induced mitophagy in renal proximal tubular cells, which was proven to protect against RIRI (Tang et al., 2018).
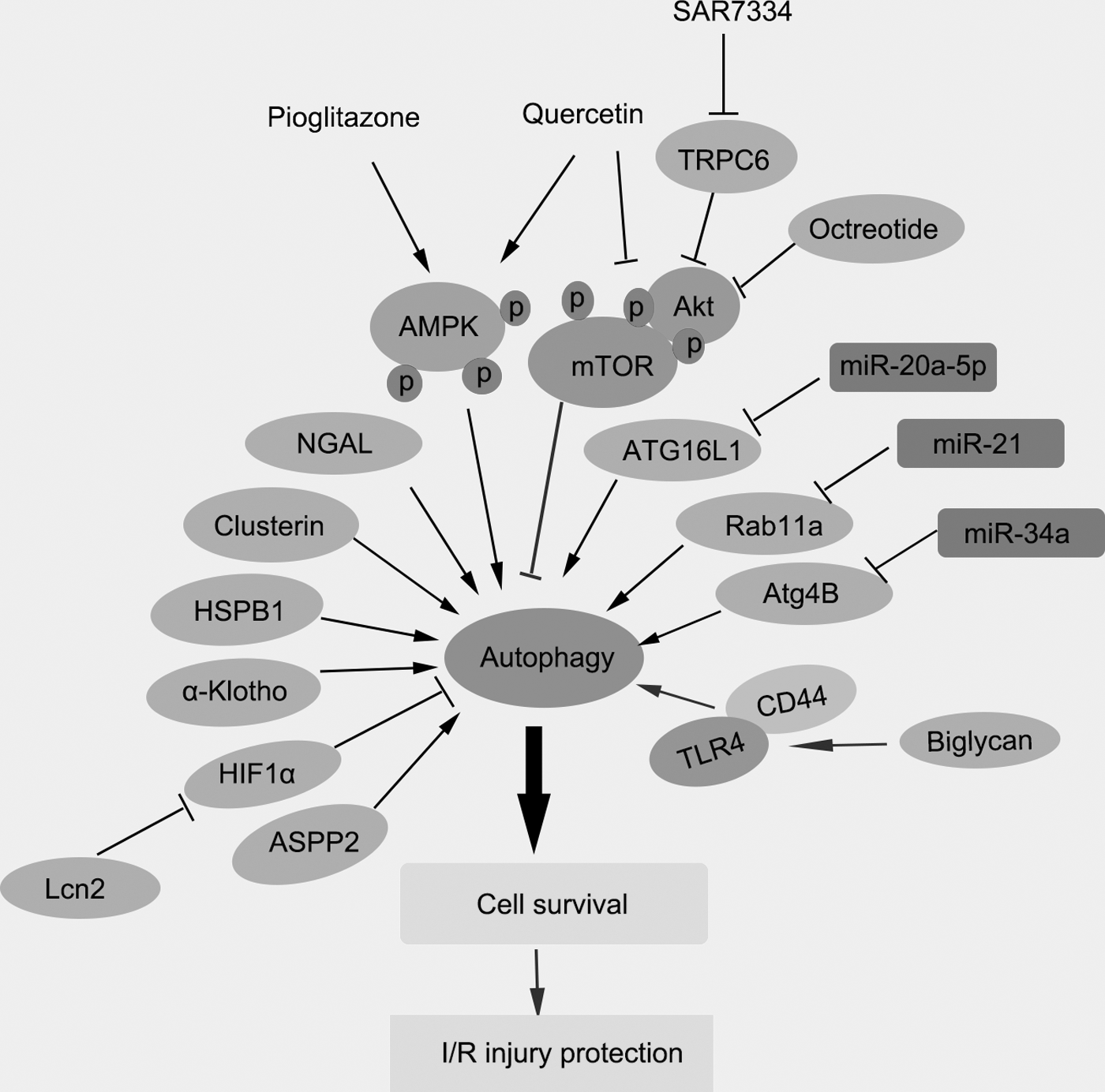
Autophagy promotes cell survival in early stage of ischemia and the regulatory agents promote cell autophagy to alleviate RIRI. Cell autophagy plays an important way in promoting cell survival in early stage of ischemia of the RIRI model. In this study, we summarize several agents, including compounds, proteins, and miRNAs, to regulate cell autophagy in ischemia/reperfusion injury progression. For instance, some agents such as HSPB1, ASPP2, AMPK, ATG16L1, Rab11a, and Atg4B induce cell autophagy to promote cell survival during RIRI model. ASPP2, apoptosis-stimulating protein two of p53; HIF, hypoxia-inducible factor; HSPB1 or HSP27, small heat shock protein beta-1; miRNAs, microRNAs; RIRI, renal ischemia-reperfusion injury; TRPC6, transient receptor potential channel 6.
In another experiment, the whole time course of autophagy was monitored after renal ischemia/reperfusion and the results revealed that autophagy occurred in a time-dependent manner, which was induced earlier than the onset of cell apoptosis (Guan et al., 2015). Moreover, activation of autophagy by rapamycin obviously alleviated renal tissue damage and tubular cell apoptosis (Guan et al., 2015). Compared with wild-type mice, telomerase knockout significantly delayed renal recovery in mice after IRI by impairing mTOR-mediated autophagy signaling (Cheng et al., 2015). Similarly, a young systemic environment effectively ameliorated IRI injuries in old mice, which was involved in decrease oxidative stress, inflammation, and apoptosis, and increased autophagy (Liu et al., 2018). Autophagy deficiency exacerbated RIRI, which was associated with higher levels of C-reactive protein (CRP), suggesting CRP was more than mere biomarker in AKI, and made the kidney more susceptible to I/R injury (Bian et al., 2017). It has been reported that hyperbaric oxygenation treatment had a protective effect in the IRI model of the transplanted rat kidneys, which was mediated by activating cell autophagy and inhibiting inflammatory responses (Bao et al., 2017). Ischemic preconditioning (IPC) promoted autophagy flux through SGK1/FOXO3a/hypoxia-inducible factor (HIF)-1alpha signaling and enhanced the protective effects in RIRI (Xie et al., 2018a). Thus, the activation of cell autophagy inhibited the occurrence of cell apoptosis, which contributed to cell survival and gave an effective protection during the RIRI process.
Excessive autophagy promotes RIRI
Although many studies revealed that autophagy played an important role in recovery from acute ischemic kidney injury, however, some articles reported that autophagy contributed to I/R-induced cell death and worsened the ischemic kidney injury (Fig. 4). For instance, inhibition of autophagy in renal tubular epithelial cells significantly inhibited H2O2-induced cell death, suggesting that high turnover of autophagy contributed to autophagic cell death during I/R injury (Suzuki et al., 2008). Several agents were reported to inhibit excessive autophagy during RIRI. For instance, fibroblast growth factor 10 (FGF10) treatment alleviated renal function and improved histological integrity in a rat model of RIRI. The molecular mechanisms clarified that FGF10 inhibited excessive autophagy by mTOR pathway and suppressed inflammatory response mediating by SQSTM1/p62 (Tan et al., 2018). Bcl-Xl transfection also improved I/R-induced renal dysfunction by downregulating the renal tubular autophagy (Chien et al., 2007). Everolimus, the inhibitor of mTOR, induced cell autophagy, which aggravated tubular dysfunction in AKI (Nakagawa et al., 2012). Exogenous zinc (Zn) worked as an effective antioxidant. ZnCl2 treatment attenuated ER stress and decreased the levels of autophagy parameters such as Beclin-1 and lysosome-associated membrane protein-2 (LAMP-2), as well as the related inflammatory responses after renal I/R (Hadj Abdallah et al., 2018). A large quantity of neutrophil gelatinase-associated lipocalin (NGAL) induced excessive cell autophagy, which promoted RIRI in AKI (Zhang et al., 2016). Briefly, during AKI, >200 ng/mL of human recombinant protein NGAL promoted the occurrence of cell autophagy in renal proximal tubular epithelial cells and the excessive autophagy increased RIRI in AKI (Zhang et al., 2016). In addition, miRNAs were reported to inhibit autophagy to protect kidney I/R injury. For instance, miR-34a exerted a protective effect in myocardial cells against IRI to inhibit cell autophagy by regulating TNFalpha signaling pathway (Shao et al., 2018). In conclusion, activation of autophagy in RIRI provided a protective effect in earlier stage of the RIRI process; however, excessive autophagy promoted cell apoptosis and cell death in the later stage of RIRI.

Excessive autophagy induces cell death and promotes RIRI. Overactivation of autophagy promotes cell death and the cell death worsens the renal I/R damage in RIRI model. Zncl2, everolimus, Bcl-XL, FGF10, or miR-34a inhibit the process of cell autophagy and the excessive autophagy promotes cell death in RIRI models. FGF10, fibroblast growth factor 10; I/R, ischemia-reperfusion; LAMP-2, lysosome-associated membrane protein-2; NGAL, neutrophil gelatinase-associated lipocalin.
The agent ameliorates RIRI through autophagy activation
As cell autophagy played an important function in cellular survival during RIRI, many agents were found to exert a protective effect by activation of autophagy in RIRI and improve the renal function during AKI. In this study, we summarized the effective agents to activate cell autophagy in IRI as well as its potential mechanisms (Fig. 3). Clear clarification of the regulators on cell autophagy in IRI might provide novel and effective therapies to prevent and treat the RIRI.
Active compounds regulate cell autophagy during I/RI
It has been reported that AMPK-regulated mTOR pathway is an important signaling pathway during autophagy induction in an in vitro I/R injury model. AMPK-induced autophagy exerted a protective function in an in vitro model of ischemia/reperfusion-induced renal tubular cell injury (Wang et al., 2013). Several compounds were reported to regulate I/R injury and protect the kidney through AMPK-mTOR signaling pathways. Quercetin, a flavonoid antioxidant, effectively promoted AMPK phosphorylation, suppressed the phosphorylation of the mTOR, and activated autophagy in the kidneys of I/R mice, suggesting quercetin could activate the AMPK-regulated cell autophagy and attenuate RIRI of I/R mice (Chen et al., 2014). Pioglitazone is a peroxisome proliferator-activated receptor (PPAR)-gamma agonist and wildly used in the therapy for type 2 diabetes. During RIRI process, pioglitazone obviously decreased the concentration of serum creatinine and urea nitrogen, improved renal histological score, and decreased the cell injury. The molecular mechanism was found that pioglitazone increased AMPK phosphorylated level, suppressed the levels of p62, inhibited the activation of caspase-3/8, and promoted the activation of LC3 II and Beclin-1 in the kidneys of IRI rats. All the data revealed that pioglitazone had a protective effect in RIRI by activating the AMPK-mediated autophagy process (Chen et al., 2018). Augmenter of liver regeneration also was reported to regulate autophagy in RIRI through the AMPK/mTOR pathway.
In some circumstances, autophagy suppressed apoptosis and played a protective role under conditions of oxidative stress (Pu et al., 2017). 22-Oxacalcitriol (OCT) treatment decreased IRI-induced upregulation of toll-like receptor 4 (TLR4), interferon gamma (IFN-g), and sodium-hydrogen exchanger-1 (NHE-1), which exerted a renoprotective effect in ischemic AKI by inhibiting cell apoptosis and promoting autophagy (Hamzawy et al., 2019). Rapamycin pretreatment obviously improved renal function, decreased inflammation response, and inhibited cell apoptosis in rat renal tissues following CIR, which was due to the induction of cell autophagy through the mTORC1/ATG13/ULK1 signaling pathway (Su et al., 2018).
Proteins regulate cell autophagy during I/RI
Some active proteins were also reported to play an important role in regulation of cell autophagy during I/R injury process. NGAL protein played a protective effect for I/R injury in male Wistar rats by improving renal function, reducing tubular cell apoptosis, increasing tubular cell proliferation, and promoting autophagy activation after RIRI (Zhang et al., 2018). Consistently, IPC was found to protect against RIRI and upregulate autophagic flux, as well as regulate the levels of serum creatinine (Scr), NGAL, and kidney injury molecule-1 (KIM-1) in renal tissues (Xie et al., 2018b). Thus, promoting the autophagy might be a novel therapy to alleviate I/R-induced AKI.
Octreotide induced autophagy and attenuated renal injury after hepatic ischemia-reperfusion (HIR) injury, which was mediated by deactivation of phosphorylation of Akt/mTOR/p70S6K signaling pathway in the kidney after HIR (Sun et al., 2017). Lipocalin-2 (Lcn2) attenuated renal injury with evidence by lower serum creatinine and inhibition of tubular epithelial cell death in mice. Importantly, recombinant Lcn2 decreased hypoxia-induced apoptosis in proximal tubule epithelial cells, and protected against RIRI through autophagy activation mediated by downregulating HIF1alpha and NF-kappaB signaling pathway, suggesting that Lcn2 could be a promising therapeutic target for clinical therapy of patients with AKI (Qiu et al., 2018). In AKI, uncoupling protein-2 (UCP2) plasmid transfection promoted autophagy and reduced cell apoptosis, which protected tubular epithelial cells in RIRI (Zhou et al., 2017). A small leucine-rich proteoglycan, biglycan, obviously increased the amounts of autophagic macrophages in mice. In addition, biglycan-CD44 interaction promoted autophagy and decreased tubular damage in IRI (Poluzzi et al., 2019). A cytoprotective protein, alphaKlotho, attenuated ischemic injury and mitigated renal fibrosis by promoting cell autophagy (Shi et al., 2016). Clusterin (CLU), a chaperone-like protein, contributed to the activation of autophagy by hypoxia in kidney cells, which was a protective mechanism for renal hypoxia/ischemia-mediated injury in hypoxic kidney tubular epithelial cells (Alnasser et al., 2016). Transient receptor potential channel 6 (TRPC6) knockdown or inhibition by a TRPC6-selective inhibitor (SAR7334) increased autophagy and inhibited apoptosis of renal tubular epithelial cells by inhibiting the PI3K/Akt/mTOR and ERK1/2 signaling pathways. Thus, TRPC6 might be a novel therapeutic target for the clinical therapy of renal oxidative stress injury (Hou et al., 2018). Apoptosis-stimulating protein two of p53 (ASPP2) deficiency inhibited cell apoptosis and enhanced autophagic flux by increasing the levels of LC3-II and p62 degradation, which effectively ameliorated AKI induced by I/R through autophagy (Ji et al., 2019). Small Heat Shock Protein Beta-1 (HSPB1 or HSP27) was reported to protect cells against oxidative stress, while in AKI model, increased HSPB1 promoted cell autophagy and inhibited cell apoptosis in renal tubular cells, indicating that HSPB1 exerted an important role in the pathophysiology of AKI (Matsumoto et al., 2015). Bcl-2/adenovirus E1B 19 kDa-interacting protein (BNIP)3 and sestrin-2, the target proteins of HIF-1alpha and p53, respectively, induced cell autophagy in renal tubules and protected the renal tubules during AKI (Ishihara et al., 2013). Knockdown of manganese superoxide dismutase (MnSOD), a major mitochondrial antioxidant, activated cell autophagy and mitochondrial biogenesis, which had a protective role in IRI following I/R (Parajuli and MacMillan-Crow, 2013). Thus, the proteins were very important to regulate the cell autophagy to protect RIRI.
miRNA regulates cell autophagy during I/RI
Cell autophagy was a key process to maintain the cellular homeostasis and exerted a protective response in ischemic kidney injury. miRNAs were reported to play an important role to regulate cell autophagy during RIRI. For instance, miR-20a-5p potentially targeted the messenger RNA (mRNA) of ATG16L1, the essential component for autophagosome formation. It has been found miR-20a-5p regulated the autophagic process through the HIF-1alpha-ATG16L1 signaling pathway in ischemic kidney injury (Wang et al., 2015). During acute ischemic kidney injury process, miR-21 level was increased and pretreatment with miR-21 inhibitor attenuated the renal injury, as accompanied by higher levels of LC3-II and beclin-1. The molecular mechanism was found that miR-21 suppressed cell autophagy by targeting Rab11a in renal I/R during renal I/R process (Liu et al., 2015a). MicroRNA-34a was also reported to suppress autophagic activity by targeting Atg4B and increased IRI in tubular epithelial cells in AKI (Liu et al., 2015b).
A dynamic hypothesis of the cell autophagy in RIRI model
The specific mechanism of cell autophagy in RIRI has not yet been clearly clarified. In this review, we summarized the role of cell autophagy in renal ischemia/reperfusion, as well as the progression of RIRI. According to the reports, under certain conditions, autophagy protects the kidneys, but in other cases, autophagy causes damage to the kidney's natural cells, which can lead to a variety of kidney diseases (Suzuki et al., 2008). In this review, we compared different studies and found that most of the researchers chose rats or mice as experimental animal models, as well as swine (Table 1). Most of the researchers chose the kidney uninephrectomized rats or mice and occluded the other renal artery and vein with a nontraumatic clamp for indicated period of time, such as 45, 30, or 120 min. As the duration of ischemic reperfusion in different studies varied with each other, the role of cell autophagy in ischemic reperfusion was also different with each other. As the pattern of autophagy in the kidney has a unique spatial and chronologic signature, whether this is protective or injurious remains controversial. Thus, a dynamic model for the protective and injurious role is predicted and summarized. In the early stage of ischemia, under moderate ischemic and hypoxic conditions, insufficient energy supply, oxidative stress, and ER stress make autophagy to be activated. Autophagy maintains the normal physiological function of cells by degrading damaged proteins and organelles, as well as producing amino acids and fatty acids. At the same time, under the condition of starvation, autophagy-degraded proteins and substrates produced by organelles can provide energy to maintain cell survival. However, cell function would be seriously worsened in some other situation, such as protracted hypoxia and the severe energy deprivation, along with a further increase in the ROS levels. At this situation, cell autophagy induces cell apoptosis to promote cell death. The tubular epithelial cells, especially the proximal tubular segment with high metabolic activity, are normally damaged by ischemia injury (Havasi and Dong, 2016).
Experimental Models of Renal Ischemia/Reperfusion
Prospects and Problems
Autophagy is a dynamic catabolic process, which is a commonly seen life phenomenon to maintain cellular homeostasis. More and more studies have confirmed that various stresses such as hypoxia, nutrient deficiencies, oxidative stress, and ER induce autophagy in kidney cells. In this review, we have summarized and discussed the role of cell autophagy in RIRI and summarized the effective agents to regulate autophagy and apoptosis in I/R injury progression. However, a few important questions remain to be clarified.
Most studies demonstrate that autophagy has protective effects on kidney cells during RIRI, such as in renal tubular epithelial cells and podocytes. However, there are also a few studies showing that excessive autophagy may aggravate kidney damage. The mechanism of action that is beneficial or harmful to the kidneys remains to be confirmed by more studies.
Second, in RIRI process, whether the cell autophagy plays a role in promoting cell survival or inducing cell death remains controversial, partly because the cell autophagy usually occurs at a certain time point. Therefore, monitoring of the whole time course of autophagy and apoptosis may help better understand the role of autophagy in RIRI.
Autophagy has not been studied in the field of traditional Chinese medicine for the treatment of kidney diseases. Therefore, it is meaningful to find effective Chinese medicine to treat kidney disease by regulating autophagy pathway and revealing its targets, which will have broad research prospects.
Authors' Contributions
J.H. selected and collected the references on the advances in cell autophagy; M.R. and W.Z. collected literature on the advances of RIRI; and J.F. and B.Y.K.L. summarized the contents and wrote the article.
Footnotes
Disclosure Statement
No competing financial interests exist.