
Abstract
Host response to viral infection is a highly regulated process involving engagement of various host factors, cytokines, chemokines, and stimulatory signals that pave the way for an antiviral immune response. The response is manifested in terms of viral sequestration, phagocytosis, and inhibition of genome replication, and, finally, if required, lymphocyte-mediated clearance of virally infected cells. During this process, cross-talk between viral and host factors can shape disease outcomes and immunopathology. Bone marrow stromal antigen 2 (BST-2), also know as tetherin, is induced by type I interferon produced in response to viral infections, as well as in certain cancers. BST-2 has been shown to be a host restriction factor of virus multiplication through its ability to physically tether budding virions and restrict viral spread. However, BST-2 has other roles in the host antiviral response. This review focuses on the diverse functions of BST-2 and its downstream signaling pathways in regulating host immune responses.
History and Molecular Characterization
Initially discovered as a surface marker for terminally differentiated and neoplastic B cells, bone marrow stromal antigen-2 (BST-2) was later reported to have diverse cellular functions (Goto et al., 1994; Ishikawa et al., 1995). BST-2 is widely expressed but its levels vary from cell to cell (Erikson et al., 2011; Hanagata and Li, 2011; Jones et al., 2012). BST-2 is a type II transmembrane protein and contains ∼180 amino acids (aas). The mature protein adopts a unique topology comprising a short N-terminal cytoplasmic tail (1–20 aas) followed by an α-helical transmembrane domain (21–48 aas), an ectodomain (49–161 aas), and a C-terminal glycosylphosphatidylinositol (GPI) domain (162–180 aas) (Kupzig et al., 2003; Hinz et al., 2010).
The cytoplasmic tail of BST-2 has a highly conserved YXY tyrosine motif known to play a role in NF-κB–mediated signaling and clathrin-mediated endocytosis (Masuyama et al., 2009; Tokarev et al., 2009; Galao et al., 2012), whereas its ectodomain contains three cysteine residues and two glycosylation sites that are highly conserved during evolution (Andrew et al., 2009). These conserved cysteine residues covalently link monomers to form dimeric or tetrameric forms of BST-2 (Swiecki et al., 2011). Two N-linked glycosylation sites (at N65 and N92) are required for the proper folding of BST-2, and the GPI domain anchors BST-2 to cell surface lipid rafts. BST-2 expression is present at the plasma membrane, as well as in the trans-Golgi network and within recycling endosomes (Hammonds et al., 2010).
Antiviral Activity of BST-2
In 2008, Bieniasz and Guatelli groups independently reported that BST-2 restricted the release of human immunodeficiency virus-1 (HIV-1). BST-2 inhibited virion release of a recombinant HIV-1 lacking the Vpu gene by tethering the nascent virion to the host cell plasma membrane (Neil et al., 2008; Van Damme et al., 2008). This prompted the name “tetherin” for BST-2. Subsequently, BST-2 was shown to tether a broad range of enveloped viruses, but its tethering activity against enveloped viruses is not a universal phenomenon. The antiviral activity of BST-2 is related to its membrane anchoring topology (Andrew et al., 2009). The two anchoring domains of BST-2 form a bridge between the budding virion and the host plasma membrane, thereby physically restricting virion release (Neil et al., 2008; Van Damme et al., 2008). Perez Caballero et al. (2009) demonstrated that the membrane anchoring domains are necessary and sufficient for viral tethering. The antiviral activity of tetherin is independent of other host factors. BST-2 transmembrane domain, dimeric ectodomain, and GPI anchor domains are critical for BST-2 antiviral activities. Tethered virions are either retained at the cell surface or mobilized for endocytic internalization and subsequent ubiquitin-based degradation (Neil et al., 2006; Miyakawa et al., 2009). BST-2 antiviral activities against a variety of enveloped viruses, including retroviruses, alphaviruses, rhabdoviruses, and mammarenaviruses, have been recently discussed in several excellent review articles (Tokarev et al., 2009; Evans et al., 2010; Arias et al., 2011; Sarojini et al., 2011; Swiecki et al., 2013; Mahauad-Fernandez and Okeoma, 2016). This review highlights other aspects of BST-2 biology, including cell signaling, immunomodulatory functions, and immunity.
BST-2 and Cell Signaling Pathways
BST-2 expression is induced by type I and type II interferons (IFNs) in response to viral infection. The role of BST-2 in cell biology was first illustrated by its potent induction of NF-κB (Matsuda et al., 2003). More recent studies confirmed a role for BST-2 in regulating NF-κB signaling (Cocka and Bates, 2012; Galao et al., 2012; Tokarev et al., 2013). BST-2 induction of NF-κB is dependent upon its multimerization or viral sensing actions (Tokarev et al., 2013). Earlier studies identified TGF beta-activated kinase 1 (TAK1) as being critical for BST-2–induced NF-κB–mediated signal transduction (Galao et al., 2012; Tokarev et al., 2013). However, subsequent studies identified additional intermediate signaling molecules with roles in activation of the NF-κB signaling pathway by BST-2. Thus, knockdown of TNF receptor associated factor 6 (TRAF6) or TNF receptor associated factor 2 (TRAF2)/ubiquitin-conjugating enzyme E2N (Ubc13) gene expression blocked BST-2–mediated NF-κB activation (Galao et al., 2012). In contrast, myeloid differentiation primary response protein MyD88, TRAF2, TAK1, TAK1-binding protein 1 (TAB1), and TAK1-binding protein 2 (TAB2) were found dispensable for BST-2–induced activation of the NF-κB signaling pathway (Tokarev et al., 2013). These findings revealed that BST-2, through induction of NF-κB signaling, can influence the host inflammatory response to a virus (Moynagh, 2005; Liu et al., 2017b), but the underlying mechanisms remain to be elucidated.
Immunomodulatory Role of BST-2 After Infection
IFNs and innate immunity
Upon pathogen encounter, pattern recognition receptors including Toll-like receptors (TLRs), Nod-like receptors (NLRs), and RIG-1–like receptors (RLRs) (Goubau et al., 2013; Cao, 2015; Chan and Gack, 2016; Liu et al., 2017a) can initiate antiviral responses, including IFN-I and proinflammatory cytokine production (Takeuchi and Akira, 2010). IFN-I signaling through the IFN receptor and JAK/STAT pathway induces expression of hundreds of IFN-stimulated genes that contribute to the establishment of an antiviral state that limits virus propagation within the infected host (Schneider et al., 2014). IFN-I responses are highly controlled and short lived, but if unchecked, excessive expression of IFNs may harm the host (Gota and Calabrese, 2003) and negatively affect hematopoiesis (Lin et al., 1998).
Plasmacytoid dendritic cells (pDCs) are among the highest producers of IFNs and inflammatory cytokines upon sensing bacterial or viral nucleic acids through TLR7 and TLR9 receptors (Colonna et al., 2004; Honda and Taniguchi, 2006). BST-2 negatively regulates the IFN-I response in pDCs (Cao et al., 2009). BST-2 is a biological ligand for the human pDC-specific receptor immunoglobin-like transcript 7 (ILT7), and binding of BST-2 to ILT7 can initiate signaling through the ILT7–FceRIy (a high-affinity IgE receptor) complex. FceRIy contains an immunoreceptor tyrosine-based activation motif in its cytoplasmic tail that mediates a calcium-dependent signaling cascade that inhibits the production of IFNs and inflammatory cytokines by pDCs (Cho et al., 2008; Cao et al., 2009). However, the detailed mechanisms underlying this negative feedback loop are largely unknown. Coculture of pDCs with BST-2-expressing cells reduced IFN-I production by pDCs after stimulation by the oligodeoxynucleotide, CpG-A (Janovec et al., 2018). Moreover, treatment with a MEK1/2 inhibitor significantly increased IFN production (Janovec et al., 2018), suggesting that the BST-2–ILT7–mediated downregulation of IFN-I is at least partially associated with MEK1/2 signaling. This pathway is highly specific for human pDCs, as ILT7 is only present in human and primate pDCs (Brown et al., 2004).
BST-2–mediated downregulation of the IFN-I response was also linked to the ability of this protein to counteract the RLR-mediated IFN-I signaling pathway (Jin et al., 2017). Specifically, BST-2 recruits the E3 ubiquitin ligase, MARCH 8, which catalyzes the lysine (K27) linked polyubiquitin chains on the mitochondrial antiviral-signaling protein (MAVS) (Jin et al., 2017), which targets MAVS for autophagic degradation through nuclear domain 10 protein 52 (NDP52) receptor. As MAVS is an essential host signaling adaptor protein for IFN-I production, its degradation negatively affected the IFN response (Jin et al., 2017) (Fig. 1). Interestingly, murine NDP52 lacks the ubiquitin-binding domain LIM-L (Thurston et al., 2009; Inomata et al., 2012; Deretic et al., 2013), which should prevent BST-2–mediated degradation of MAVS. In fact, pDCs from BST-2 knockout mice showed reduced IFN-I secretion in response to viral challenges (Swiecki et al., 2012). These results suggest that evolutionary selection of the LIM-L domain in NDP-52 and ILT7 expression by human pDCs cells influences how BST-2 regulates IFN-I production in these cells relative to mouse pDCs. Additional research is required to determine how BST-2 affects the innate antiviral immune response in different species.

BST-2 regulation of the IFN-I response. (1) Virion interacts with cell surface receptors to enter the cell. (2) Viral genome (RNA) is recognized by the RLRs. RIG-I signals are transduced to the transcription factors through stimulation of MAVS at the mitochondrion-associated membrane. Activation of MAVS leads to phosphorylation of IRF3. Phosphorylated dimers of IRF3 then translocate to the nucleus where they bind and activate specific promoters triggering expression of IFNs. (3) Type-I IFNs interact with IFNAR, recruit, and phosphorylate the STAT1 and STAT2. STAT1 and STAT2 form a heterodimer that, in turn, recruits the IRF9 to make a complex. This complex translocates to the nucleus and induces expression of genes (e.g., BST-2) regulated by IFN-stimulated response elements. (4) BST-2 recruits the E3 ubiquitin ligase MARCH 8. (5) MARCH 8 then catalyzes the K27-linked polyubiquitin chains on MAVS at K7 position. (6) Cargo receptor NDP52 recognizes ubiquitinated MAVS. (7) NDP52 delivers MAVS to autophagosome for degradation. (8) BST-2-mediated autosomal degradation of MAVS and terminal of RIG-I, MAVS-mediated IFNI production via a negative feedback manner. BST-2, bone marrow stromal antigen 2; IFN, interferon; IRF3, IFN response factor 3; IFNAR, IFN-α/β receptor; MAVS, mitochondrial antiviral-signaling protein; RLR, RIG-1–like receptor; STAT, signal transducers and activators of transcription.
Adaptive immunity
Although BST-2 has conserved coding regions, polymorphic forms do exist across species. Thus, NZW/LacJ (NZW) mice contain BST-2 alleles lacking the endosomal-sorting motif (YxY) and show higher BST-2 cell surface expression than C57BL/6 mice, whose BST-2 contains the YxY motif. Consistent with its higher cell surface expression, BST-2 from NZW mice exhibited a more potent antiviral activity against Friend murine leukemia retrovirus (F-MuLV) than BST-2 from C57BL/6 mice (Barrett et al., 2012). However, the endocytosis-competent version of BST-2 in C57BL/6 mice showed a greater ability to control viremia, suggesting a role for immune modulatory functions linked to BST-2 (Li et al., 2014). Consistent with this hypothesis, enhanced restriction of F-MuLV in C57BL/6 mice was associated with a stronger IFNγ response in NK cells, CD4+ T cells, and CD8+ T cells (Li et al., 2014). Increased endocytosis of virions by pDCs in C57BL/6 mice could trigger TLR3-mediated IFN-I production, leading to augmented NK function, as these cells are highly responsive to TLR3- and TLR7-dependent cytokine stimuli (Swiecki et al., 2012; Gibbert et al., 2014; Li et al., 2014, 2016) (Fig. 2A). This might account for lower IFN-I production observed in BST-2–deficient pDCs (Swiecki et al., 2012). Because BST-2 can influence IFN-I levels, it has the potential to modulate host defense during both the early and late phases of viral infection. Accordingly, using the mouse model of chronic infection by lymphocytic choriomeningitis virus (LCMV), we showed that the early confinement and sequestration of virions in the splenic marginal zone were compromised in the absence of BST-2 (Urata and Kenyon, 2018). This resulted in alterations in antiviral T cell priming, leading to reduced T cell proliferation and effector functions (e.g., IFNγ and TNFα). Peripheral control of a chronic LCMV infection was also compromised in BST-2–deficient mice, and the virus established long-term persistence in the brain (Urata and Kenyon, 2018). These findings illustrated how BST-2 can influence both innate and adaptive immune responses to viral infections. Further studies are required to understand the direct versus indirect contributions of BST-2 to antiviral immunity, and how the functionality of this protein can be enhanced to help fight infections.

Antiviral and immunomodulatory functions of BST-2.
The relationship between BST-2 and antibody-dependent cellular cytotoxicity (ADCC) in HIV-1–infected cells is well established (Arias et al., 2014). Higher surface expression of Env in infected cells could facilitate ADCC. HIV-1 mutants with impaired Vpu-mediated BST-2 counteracting activity show increased surface accumulation of Env protein in infected cells, which can facilitate ADCC. Thus BST-2 increases the susceptibility of Vpu-mutated HIV-infected cells to ADCC and knockdown of BST-2 decreases the sensitivity of HIV-infected cells to ADCC (Arias et al., 2014). Notably, overexpression of BST-2 in response to type I IFN, interleukin (IL)-27 or by other means, upregulates surface expression of Env protein and further stimulates HIV-1–infected cells for ADCC-mediated elimination (Pham et al., 2016; Richard et al., 2017) (Fig. 2B). These studies demonstrate an indirect connection of BST-2 and ADCC phenomena in HIV-1–infected cells, although this phenomenon is not yet reported in other viral infection models. Therefore, BST-2 could modulate interplay between innate and adaptive immune response to control viral replication.
Other immunomodulation roles of BST-2
Various other immunomodulation functions of BST-2 have also been reported. For example, in addition to tethering of viral envelope, it also reported to tether exosomes and could influence its cellular target and interactions (Edgar et al., 2016). Exosomes are involved in host immune responses and linked with various diseases such as cancer and neurodisorders (Soria et al., 2017; Tai et al., 2018). In case of cancer, exosome carries damage-associated molecular patterns (DAMPs) in myeloid cells that stimulate inflammatory cytokine productions and could promote cancer progression (Hoshino et al., 2015; Nabet et al., 2017). Alternatively, tumor exosomes could also suppress innate antiviral immune response by transferring the activated epidermal growth factor receptor (EGFR) to the dendritic cells (Gao et al., 2018) (Fig. 2C). Thus BST-2's tethering action on exosome could influence outcome of host immune response to tumor. In a different model, Sally James et al. identified a new subset of nondifferentiating bone marrow stromal cells (BMSCs), which exclusively expresses higher BST-2 and IL-7, although specific functional roles of BST-2 in these BMSCs are yet to be defined (James et al., 2015). A similar observation was made wherein higher cells surface expression of BST-2 was noticed in cancellous bone fragments resident cells (El-Sherbiny et al., 2018). However, relevance of BST-2 expression and its mechanistic role is yet to be elucidated.
BST-2 and Cancer
Increased BST-2 expression has been documented in different cancer tissues, including ovarian, lung, head and neck, cervical, thyroid, breast, endometrial, pancreatic glioblastoma, and myeloma (Wang et al., 2009; Wainwright et al., 2011; Tai et al., 2012; Yokoyama et al., 2013; Fang et al., 2014; Mahauad-Fernandez et al., 2014, 2015; Milutin Gasperov et al., 2014). BST-2 overexpression at the early stage of multiple myeloma suggested that BST-2 might be a suitable target for cancer immune therapy. However, not all cancer types exhibit increased BST-2 expression. BST-2 expression is unchanged in thyroid and lung adenocarcinomas and is downregulated in liver, kidney, lung squamous, and prostate cancer relative to normal cells (Mahauad-Fernandez et al., 2015). Almost all breast tumors express BST-2 to a certain level, and a higher expression level of BST-2 is associated with aggressive and progressive malignancy (Mahauad-Fernandez et al.,
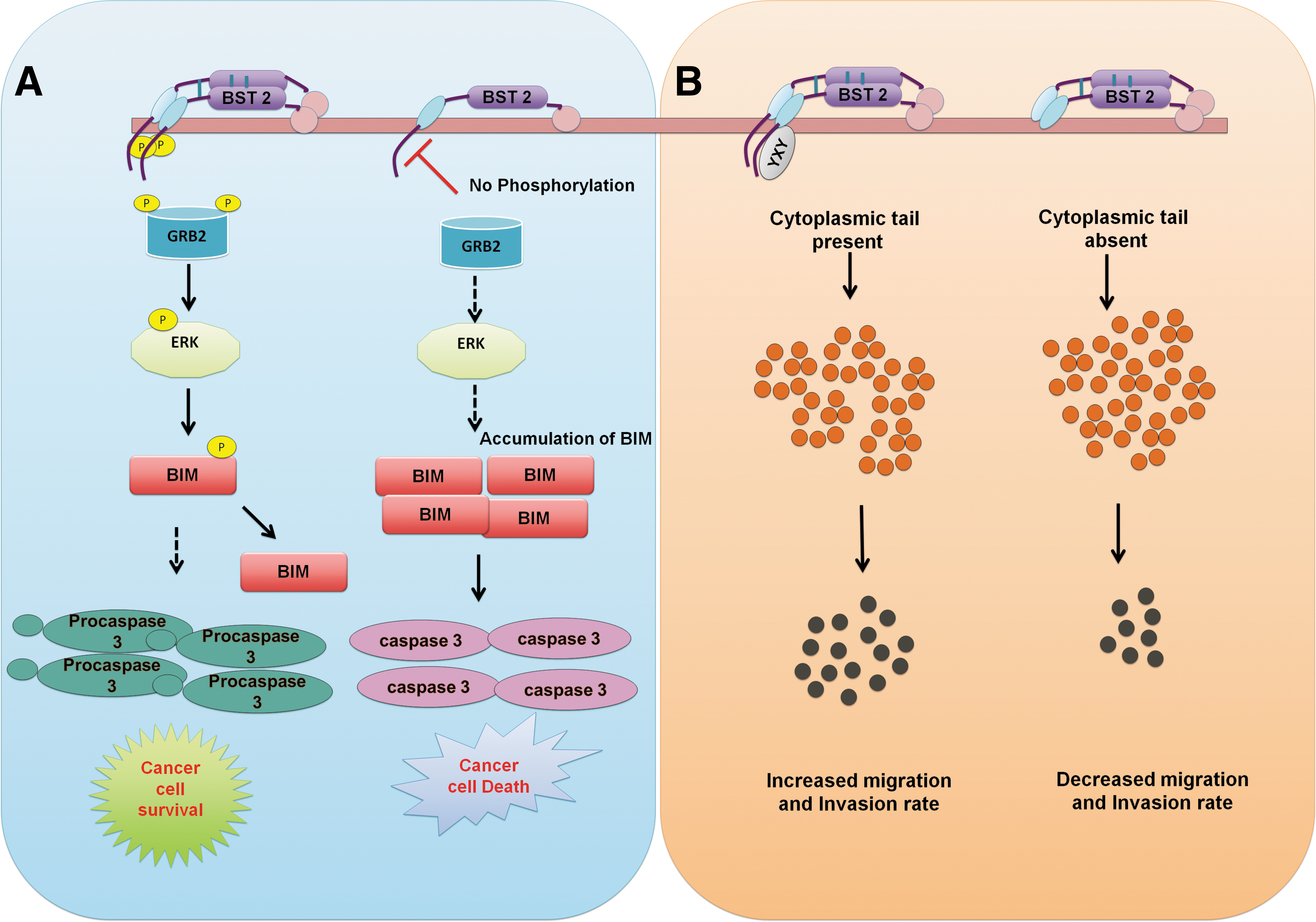
The influence of BST-2 on tumor cell survival, invasion, and migration.
Conclusion
Cells have an array of factors that play precise roles in maintaining normal physiology and responding to diverse challenges (infections, tumors, etc.). A plethora of factors have been discovered that contribute to host defense against pathogens and the maintenance of immune homeostasis. BST-2 is one such factor, and studies have highlighted multifaceted biological roles for BST-2 in antiviral responses, cell signaling, immune modulation, and even malignancy. Because the immune system has the potential to cause pathology, it is important that responses are precisely regulated. A role for BST-2 in immune modulation is supported by its ability to alter IFN-I, NF-κB, and T cell effector functions. The detailed mechanisms by which BST-2 executes these functions remain to be elucidated, but future studies are likely to uncover a broader role for BST-2 in shaping host responses to infection and cancer. This knowledge may facilitate the development of therapeutics based on regulating BST-2 functionality.
Footnotes
Authors' Contributions
R.T. and D.N. structured the article and gave their inputs toward shaping the article. J.C.d.l.T. and D.B.M. made critical suggestions and editing the article.
Disclosure Statement
No competing financial interest exists.
Funding Information
R.T. is thankful to University Grant Commission (UGC), Govt. of India, for his graduate fellowship.