
Abstract
Long noncoding RNA (lncRNA) was closely attached to various cancers according to previous studies. In this study we aimed to investigate an lncRNA signature with prognostic value of over survival (OS) outcomes of gastric cancer (GC). Profiles of mRNAs expression and clinical information of 381 GC tissues and 32 nontumor gastric tissues were downloaded from The Cancer Genome Atlas database. Comparison of various lncRNA expression between cancer tissue and normal tissue was made among these data. In the end, a nine-lncRNA signature was discovered using univariate and multivariate Cox regression analyses, with a prospect possibility of the OS in GC patients. Receiver operating characteristic (ROC) was used to evaluate the accuracy of survival model. The gene ontology and Kyoto Encyclopedia of Genes and Genomes pathway enrichment analyses were used to predict the possible functions and pathways of these lncRNAs. Altogether 720 distinctively expressed lncRNAs were selected between GC and normal tissues. By univariate and multivariate Cox regression analyses, nine lncRNAs were eventually filtered to set a predictive model, distributing patients into high-risk and low-risk groups with extraordinary different OS. Area under the curve of the ROC curve for the nine-lncRNA signature's prediction of 5-year OS was 0.795. Further functional enrichment analyses indicated that these lncRNAs may be associated with biological processes such as protein binding, DNA replication, and cell cycle. Our study identified a nine-lncRNA signature, which could act as a potential prognostic biomarker in the prediction of GC patients' OS.
Introduction
Gastric cancer (GC) is a universally fatal disease with >1,000,000 new cases diagnosed in 2018 and an estimated 783,000 deaths (Bray et al., 2018). Despite improvements in the diagnosis and therapy of this disease in the past decades, a significant number of patients were still diagnosed in late stage with extremely poor survival rate. Recent studies have proved that the criteria alone such as the tumor, node, metastasis (TNM) stage, where a tumor can be grouped into various stages according to different TNM, is not sufficient for estimating prognosis (Bornschein et al., 2011). Therefore, it is imperative to specify novel biomarkers, which could predict patients' over survival (OS) time and be used as therapeutic targets.
Long noncoding RNA (lncRNA) is transcribed molecules ≥200 nucleotides in length (Gloss and Dinger, 2016), playing an important role in various biological processes, such as chromatin remodeling, cell differentiation, and carcinogenesis (Gibb et al., 2011). Accumulating studies have identified that lncRNA might be closely associated with tumor genesis, including lung cancer (Zhang et al., 2018a; Liu et al., 2019), liver cancer (Yuan et al., 2017; Ma et al., 2019), breast cancer (Li et al., 2018; Beltran-Anaya et al., 2019), ovarian cancer (Hosseini et al., 2017), colorectal cancer (Ma et al., 2016), prostate cancer (Mehra et al., 2016), and esophageal cancer (Huang et al., 2018). Furthermore, diversification of lncRNA is associated with tumor proliferation, invasion, and metastasis of various types of cancers (Ma et al., 2018). Therefore, lncRNAs might be potential diagnostic biomarkers for cancer.
Many scholars have studied the expression of lncRNA in GC and found that many lncRNAs were aberrantly expressed in GC tissues, including lncRNA GClnc1 (Sun et al., 2016), lncRNA GMAN (Zhuo et al., 2019), and lncRNA HOXC-AS3 (Zhang et al., 2018b). As the expression of lncRNA is relatively low, bias exists if one single lncRNA is regarded as a biomarker. Fortunately, a combination of diverse potential lncRNA biomarkers is deemed to have advantages (Wu et al., 2018). In addition, RNA sequencing (RNA-seq) is recently used for transcriptome analyses with great popularity (Han et al., 2015; Arrigoni et al., 2016). And the development of this method showed more complete characterization (Ozsolak and Milos, 2010). Consequently, herein we develop a novel nine-lncRNA–related prognostic model from the 413 GC patients' RNA-seq samples in The Cancer Genome Atlas (TCGA) data set, whose survival-related risk score could serve as a potential prediction of the OS in GC patients.
Materials and Methods
Patient data
The mRNA expression of stomach adenocarcinoma (STAD) (Supplementary Appendix) study of the TCGA program was obtained from the publicly available TCGA database * up to January 24, 2019. A total of 413 RNA-Seq were downloaded, including 381 gastric tumor tissues and 32 cases of gastric tissue adjacent to tumor. Corresponding clinical information, including age, gender, vital status, race, TNM stage, and histological grade, was also obtained and assessed. Totally 386 GC patients were recruited to our study (Table 1). All the data could be reached freely on the internet. Hence there is no need for approval by the ethics committee.
Summary of Clinical Characteristics of Gastric Cancer Patients
NA, MX, GX: clinical data are unknown.
Differentially lncRNA expression level
The gene expression level of the 381 GC tissues and 32 gastric tissues adjacent to the tumor was collected from the TCGA database. The edgeR package in R language was applied to analyze different expression levels of lncRNA in each sample. And |log2(fold change)| ≥1 and p < 0.05 were identified as effective for the next studies.
Overall survival analysis
The relationship between each feature's expression level and OS was assessed by univariate Cox regression adopting a survival R package. And lncRNAs with p < 0.01 were entered into the next multivariate Cox regression process. Afterward, an lncRNA-related prognostic model was built up to assess each patient's survival risk as the following:
where N refers to prognostic lncRNA number, Ci suggests the ith lncRNA coefficient, and Ei denotes the ith lncRNA expression level. And then a concise forest map is drawn based on the model obtained by multivariate regression analysis.
Risk stratification and ROC curve
The model equation for risk assessment was used for every patient, and 374 patients were split into low- and high-risk groups according to the average score. OS curves were manufactured applying the Kaplan–Meier method, and the log-rank test comparison was utilized between the mentioned two groups.
To compare the expression of nine lncRNAs in different risks, we drew a heat map using R package of “pheatmap.” Area under the curve (AUC) of the ROC indicated the ability of the lncRNA prognostic model calculating by a “survival ROC” package. And concordance index (C-index) check using the R package of “survcomp” was added to verify the AUC value.
Functional enrichment analysis
To judge biological course referring to the predictive lncRNA, functional enrichment analysis was performed. Expression levels of the nine lncRNAs and each protein coding genes (PCGs) were calculated by the means of Pearson correlation coefficients. And 2979 genes were significantly considered to be lncRNA-related PCGs with |Pearson correlation coefficient| >0.40.
Results
Patient characteristic
In this study, a total of 413 GC RNA-seq expression profiles were downloaded from the TCGA data portal, which includes 381 samples of GC tissue and 32 samples of gastric tissue adjacent to the tumor. These samples were collected from 386 patients with GC and 32 adjacent tissues. Among which, 27 patients with GC and adjacent tissues were matched: 354 patients with only GC and 5 patients with only adjacent tissues. And their clinical and pathological characteristics are illustrated in Table 1.
Differentially expressed lncRNA between GC and normal tissues
Based on the screening criteria [|log2 (fold change)| ≥1 and p < 0.05], totally 720 differentially expressed lncRNAs were investigated between GC and normal tissues, involving 540 (75.0%) upregulated and 180 (25.0%) downregulated (Supplementary Appendix). A volcano plot (Fig. 1) was displayed for the purpose of clarifying differentially expressed lncRNAs between the two groups.
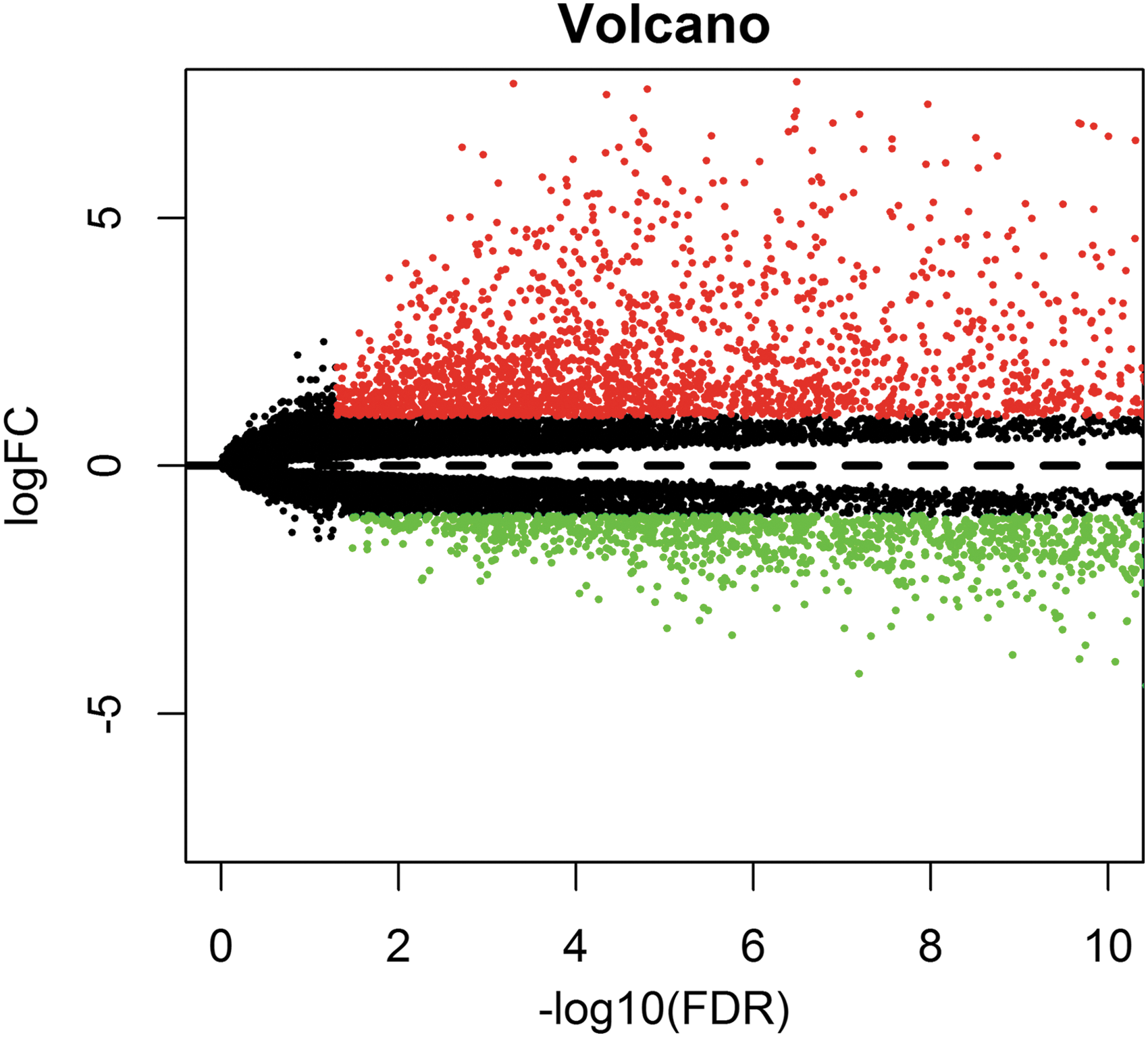
Volcano plot of differentially expressed lncRNAs. Green dots signify downregulated lncRNAs, whereas red dots imply upregulated lncRNAs and black dots represent lncRNA with no differential expression. lncRNA, long noncoding RNA.
Prognostic assessment estimate
The relationship between differentially expressed lncRNAs and patient's OS was calculated employing a univariate Cox model. Totally 16 lncRNAs were screened out as candidate biomarkers for GC patients (p < 0.01) (Table 2). For 16 lncRNAs selected by single-factor Cox analysis, multifactor Cox analysis was performed, and Akaike information criterion values were filtered, and genes with similar expression levels were excluded. Finally, the optimal model of nine lncRNAs was established. And the detailed information of nine lncRNAs is illustrated in Table 3.
Long Noncoding RNA Predictors by Univariate Cox Analysis
p < 0.01.
HR, Hazard ratio; lncRNA, long noncoding RNA.
Detailed Information of Nine lncRNAs for Gastric Cancer Patients
p < 0.01.
Coef, coefficient.
We performed a risk-score formula as the predictive model for OS prediction based on the expression levels of the nine lncRNAs applying the multivariate Cox regression coefficient as follows: (0.1241 × expression level of ADAMTS9-AS1)+(0.1395 × expression level of LINC01614)+(−0.1323 × expression level of LINC01210)+(0.1032 × expression level of OVAAL)+(0.1063 × expression level of LINC02408)+(0.1070 × expression level of FLJ42969)+(−0.2810 × expression level of LINC01775)+(0.0840 × expression level of LINC01446)+(0.1141 × expression level of CYMP-AS1).
Hazard ratio of nine lncRNAs is shown in a forest map. Among these nine lncRNAs, seven were illustrated with high risk (ADAMTS9-AS1, LINC01614, OVAAL, LINC02408, FLJ42969, LINC01446, CYMP-AS1, coefficient >0), whereas two were suggested to be protective (LINC01210, LINC01775, coefficient <0). The overall p-value is 2.777E-11, and the C-index value of the model is 0.68 (Fig. 2).
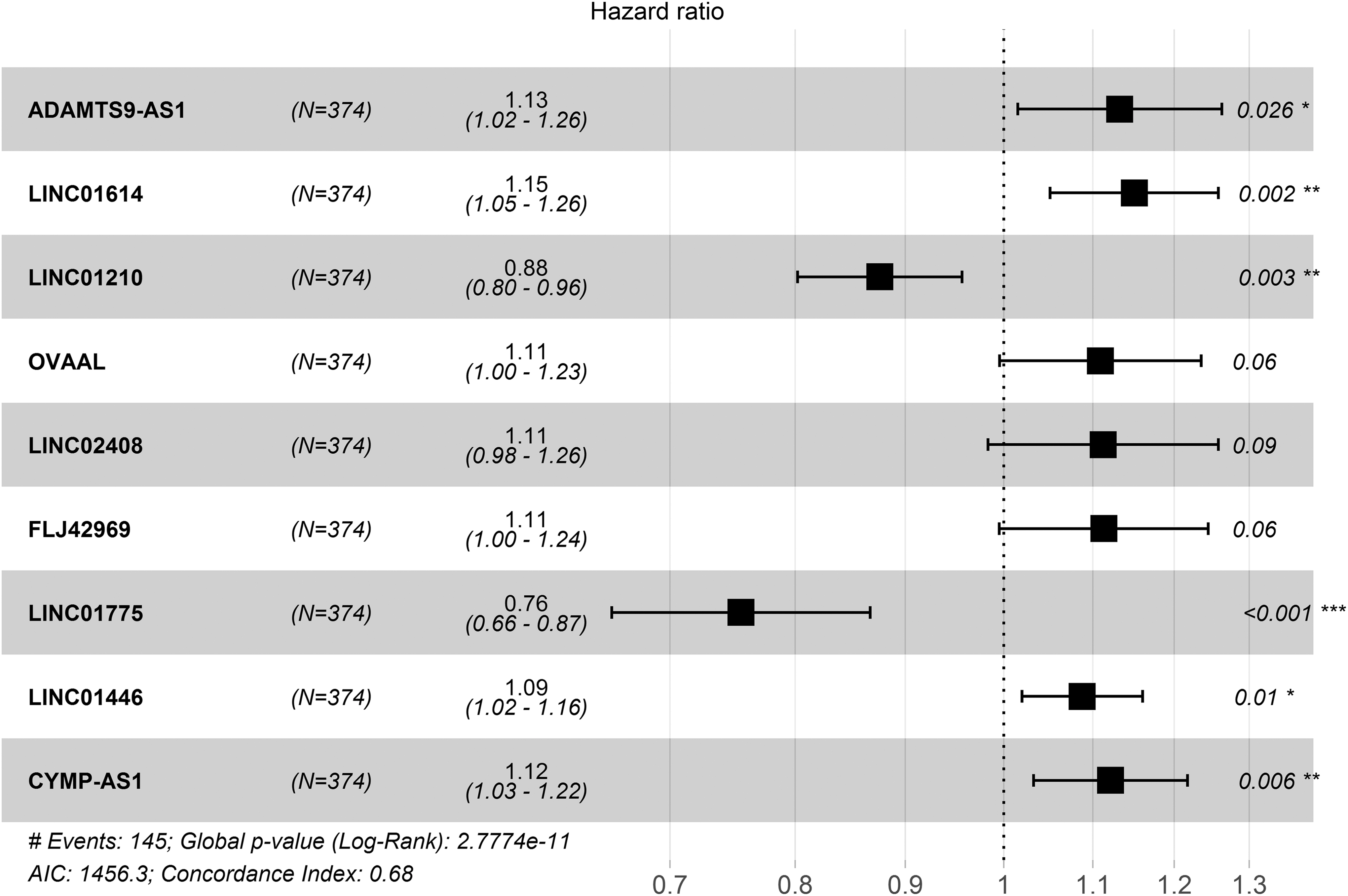
Forest plot for the association between nine lncRNAs and risk value. *p < 0.05, **p < 0.01, ***p < 0.001.
Risk stratification and ROC curve
Patients who have lost follow-up were excluded in our study. For the remaining 374 patients, we assessed each patient's survival risk score according to the expression levels of the nine lncRNAs. Then we ranked the risk scores of the patients, and stratify subjects into high-risk and the low-risk groups exploiting the average risk score as the cutoff point (Fig. 3A).

LncRNA predictive risk score analysis of 374 GC patients.
After that, we found people with increasing risk score tended to have worse survival status (Fig. 3B). And the nine-lncRNA expression levels were shown in a heat-map manner, in which the pink bar means high expression and the green bar means low expression (Fig. 3C).
The Kaplan–Meier curve suggested that patients in the low-risk group have prolonged OS and disease-free survival time compared with the patients in high-risk group (p < 0.001) (Fig. 4A, B). Area under ROC curve of the nine-lncRNA signature was 0.795, revealing that the nine-lncRNA prognostic model plays an effective role in the prediction of the OS among GC patients (Fig. 4C). The model is indeed accurate as the C-index value is 0.6810 (95% CI: 63.3–72.9%, p < 1.3525E-13).

Kaplan–Meier and ROC curve for the nine-lncRNA signature of GC patients.
Functional assessment of the nine lncRNAs
We carried out functional enrichment analyses (gene ontology [GO] and Kyoto Encyclopedia of Genes and Genomes [KEGG]) for the PCGs coexpressed with the prognostic nine lncRNAs. GO functional annotation suggested that among 2979 coexpressed mRNAs with nine lncRNAs, 65 GO terms were significantly enriched. The top 30 GO terms are signified in Figure 5A, which majorly clustered in regulation of various biological processes, such as GO:0019838∼growth factor binding, GO:0010628∼positive regulation of gene expression, GO:0015631∼tubulin binding, GO:0005516∼calmodulin binding. KEGG analysis focusing on the biological pathways justified that 2979 coexpressed mRNAs were enriched to 31 KEGG pathways. The top 30 pathways are illustrated in Figure 5B, which mainly consist of hsa03030: DNA replication, hsa04020: calcium signaling pathway, hsa04110: cell cycle. All in all, these results expressed that nine lncRNAs may be touched upon the tumor with protein binding, DNA replication, and cell cycle.
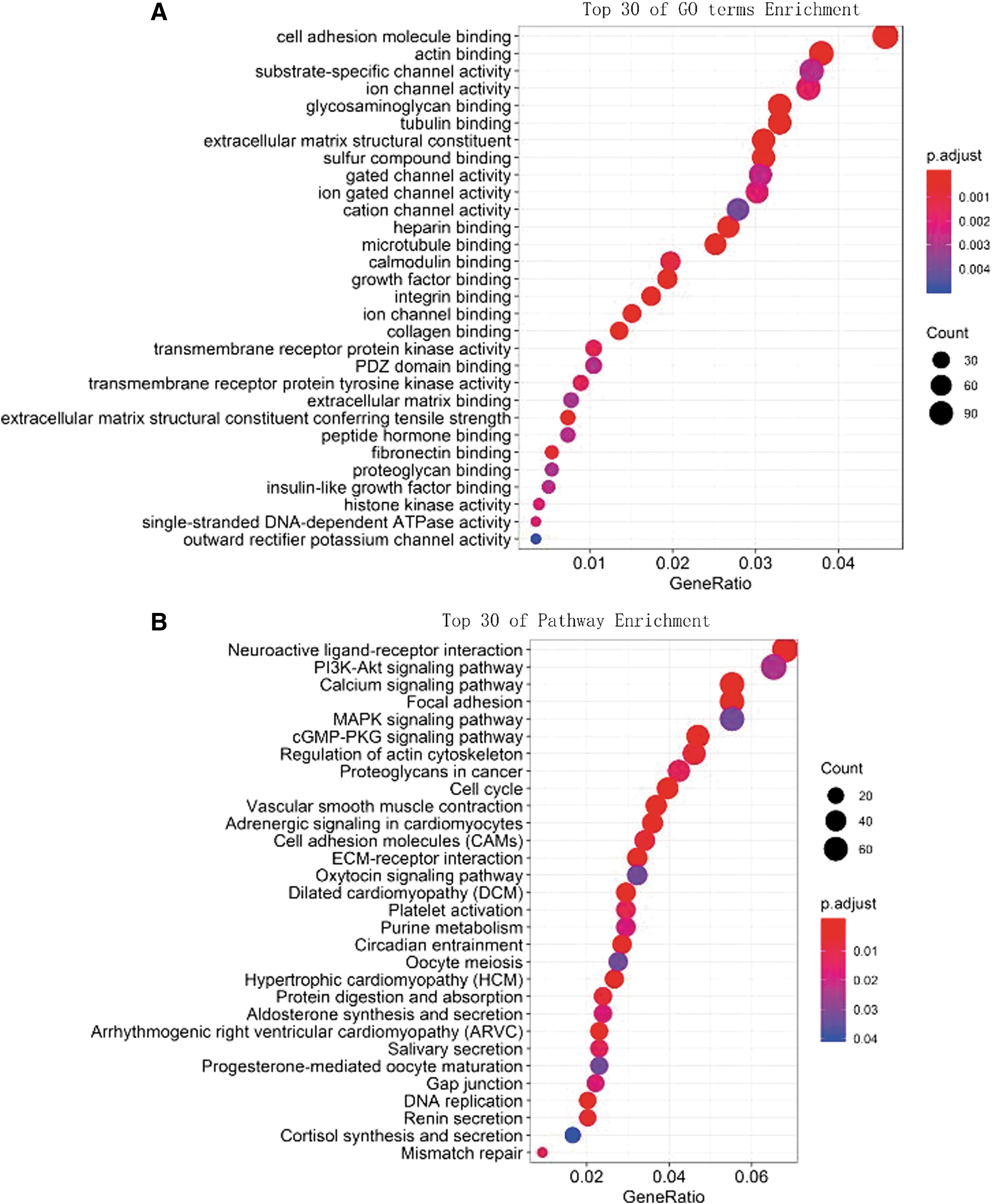
Top 30 enrichments of
Discussion
Before the discovery of noncoding RNAs, researches for cancer were mostly focused on PCGs (Zhang et al., 2018b). Contemporary expanding evidence accumulated that lncRNA might be closely associated with cancer biogenesis and prognosis (Song et al., 2018). Sun et al. (2016) showed that lncRNA GClnc1 was upregulated and related to tumor genesis, tumor size, metastasis, and poor prognosis in GC. Lin et al. demonstrated that the aberrantly lower expression of lncRNA metallothionein 1D, pseudogene (MTM) in GC is remarkably associated with metastasis. Consequently, lncRNAs could serve as biomarkers in GC.
As is known to all, cancer has complex molecular characteristics. Therefore, single lncRNA expression pattern may not be enough for the exact prediction of GC prognosis. Fortunately, as previous studies have shown, combination of myriad potential lncRNA biomarkers could improve the prediction accuracy (Wu et al., 2018). Ke et al. (2017) demonstrated that four lncRNAs (AK001058, INHBA-AS1, MIR4435-2HG, and CEBPA-AS1) could act as a biological marker for GC patients. Zheng et al. (2019) determined three lncRNAs (FAM49B-AS, GUSBP11, and CTDHUT) could be used for diagnosing GC. The mentioned researches were all analyzed based on microarray technology. But this technology has some disadvantages, such as bias of selection and limitation of spotting transcripts (Farkas et al., 2015; Han et al., 2015).
Currently a novel technology named RNA-seq technique is catching more and more attention due to its benefit reducing the mentioned shortcomings (Farkas et al., 2015). Nevertheless, research based on RNA-seq technology inspecting the effect of lncRNA on GC patient OS is quite deficient. As far as we are concerned, our study is the first lncRNA-associated predictive model using RNA-seq technology. In our study, a novel nine-lncRNA signature was displayed as an absolute prognostic impact on OS for GC patients. Furthermore, ROC illustrated that the AUC of the nine lncRNAs was 0.795. These results demonstrate that these nine lncRNAs work as a crucial target both in predicting the OS time of GC patients and in therapy. This could benefit clinical doctors from selecting more suitable treatments for patients with different survival risks.
Despite the popularity of lncRNA, the functions of most lncRNAs are still ambiguous. Many studies have shown that lncRNA affects cell replication and cell cycle of GC through a variety of pathways. Liu et al. (2018) demonstrated that lncRNA HNF1A-AS1 contributed to GC progression through modulation of the cell cycle. Pan found that LncRNA differentiation antagonizing non-protein coding RNA (DANCR) knockdown inhibited the proliferation of GC cells by inducing cell cycle arrest and cell apoptosis (Pan et al., 2018). And analysis on lncRNA functions involved in coexpressed mRNAs has been validated to be efficacious (Liao et al., 2011). For the purpose of studying the functions of the nine lncRNAs, the relationship between their expression levels and the coexpressed PCGs was explored. We conducted GO and KEGG enrichment analysis. Our results show that these lncRNAs may regulate genes through protein binding, DNA replication, and cell cycle, resulting in gastric carcinogenesis.
Some imperfection of our study should be taken into consideration. First, the prognostic ability of the nine-lncRNA model was merely accurate based on the TCGA data. And this database mostly included the race of white Americans. We checked ICGC, TARGET, and other available databases to strengthen our discovery, but no suitable GC RNA-seq was found. Second, we did not analyze some conventional clinical factors such as race, gender, age, and tumor stage, which might have an effect on predicting OS outcomes of GC patients. Third, the molecular mechanism of these lncRNAs remains unclear, which need to be investigated in-depth in the future.
Conclusions
In summary, our study certificated a nine-lncRNA signature which might become a novel prognostic indicator for the survival risk of GC patients. Functional predictions revealed that the nine-lncRNA signature is relevant to protein binding, DNA replication, and cell cycle. Further studies are needed to analyze the association with other clinical parameters and a possible clinical application of the nine-lncRNA signature.
Footnotes
Authors' Contributions
C.C., L.Y., and Y.T. analyzed and interpreted the data, and drafted the article together. H.W., Y.H., and H.J. contributed to the acquisition of data and collection of relevant literature. K.Z. conceived the study, participating in its design and coordination.
Disclosure Statement
No competing financial interests exist.
Supplementary Material
Supplementary Appendix
References
Supplementary Material
Please find the following supplemental material available below.
For Open Access articles published under a Creative Commons License, all supplemental material carries the same license as the article it is associated with.
For non-Open Access articles published, all supplemental material carries a non-exclusive license, and permission requests for re-use of supplemental material or any part of supplemental material shall be sent directly to the copyright owner as specified in the copyright notice associated with the article.