
Abstract
Endogenous phytohormones auxin (indole-3-acetic acid [IAA]), abscisic acid (ABA), gibberellin (GA3), and brassinosteroid (BR) play a role in responses to drought stress in higher plants. Tea plant is one of the major economic corps worldwide. The tender shoots of tea plants are the main source for tea production. The effects of drought stress on endogenous IAA, ABA, GA3, and BR metabolisms in tender shoots of tea plants need to be illustrated. In this study, a total of 17 IAA-related genes, 17 ABA-related genes, 18 GA3-related genes, and 8 BR-related genes were identified under drought stress in tender shoots of tea plants, respectively. By using a combination of phytohormone determination, phylogenetic tree construction and sequence analysis, gene expression profiles, functional classification, Kyoto encyclopedia of genes and genomes enrichment, and distribution of genes analysis, we have demonstrated that IAA, ABA, GA3, and BR metabolisms might participate in the regulation of the response to drought stress in tender shoots of tea plants. The expression level of CsLYCE negatively correlated with ABA accumulation under drought stress. Our findings could shed new light on the effects of drought stress on the IAA, ABA, GA3, and BR metabolisms in tender shoots of tea plants.
Introduction
Tea plant [Camellia sinensis (L.) O. Kuntze] is a small tree or evergreen shrub that belongs to the genus Camellia of Theaceae. Tea is an important economic crop worldwide (Chen et al., 2009). The tender shoots are the major source of tea, which is a popular beverage in the world. Based on the Food and Agriculture Organization of the United Nations using imputation methodology, * the harvested area of tea plants in China was estimated at 2,212,750 hectare (ha) in 2017. “Longjing 43” and “Longjingchangye” are the tea plant cultivars with high quality and adapt to abiotic stress environment in Eastern China. “Longjing 43” and “Longjingchangye” are widely used for genetic analysis and research of tea plants (Wang et al., 2016; Li et al., 2018).
Drought stress has severely limited the growth and development of higher plants (Shao et al., 2009). The amount of phytohormones and the expression levels of phytohormone-related genes could be affected by drought stress in higher plants (Tamiru et al., 2015; Abid et al., 2017). Drought stress can induce the damages in growth and quality of tea shoots (Zhou et al., 2014). Phytohormones are closely associated with plant growth, development, abiotic stress tolerance, and productivity. Phytohormones could increase the resistance to abiotic stress in higher plants (Ahmad et al., 2010). Abscisic acid (ABA) could enhance drought stress tolerance in wheat seedlings (Wei et al., 2015). Auxin (indole-3-acetic acid [IAA]) plays a vital role in responding to oxidative damage in higher plants (Krishnamurthy and Rathinasabapathi, 2013). Gibberellin (GA3) could improve salt stress tolerance in soybeans (Hamayun et al., 2010). Brassinosteroid (BR) also plays an important role in responding to harsh environmental conditions (Ye et al., 2017).
Transcriptome analysis suggests that endogenous phytohormones are involved in the developmental processes of rice microspore/pollen (Hirano et al., 2008). Phytohormones are involved in endodormancy release in flower bud transitioning of Japanese pear (Pyrus pyrifolia Nakai) (Bai et al., 2013). The biosynthesis and signaling of phytohormones play a pivotal role in the sex differentiation of Castanea henryi flower (Fan et al., 2017). The genes related to phytohormones are differentially expressed in soybean leaves under drought stress (Le et al., 2012).
In this work, to assess the effects of drought stress on metabolism of IAA, ABA, GA3, and BR, the genes involved in metabolism of IAA, ABA, GA3, and BR in drought-induced tea shoots were identified. The levels of IAA, ABA, GA3, and BR, phylogenetic tree, gene sequence, expression profiling, functional classification, pathway enrichment, and gene distributions were analyzed. Our work presented a strategy on illustrating the potential roles of IAA, ABA, GA3, and BR metabolisms in tea shoots under drought stress.
Materials and Methods
Plant materials, growth conditions, and drought stress treatment
One-year-old cutting seedlings of tea plant cultivar “Longjing 43” were used as plant materials. The original source of the plant materials used in this study was acquired from the Tea Science Research Institute, College of Horticulture, Nanjing Agriculture University, Nanjing, China (32°02′ N, 118°50′ E). The tea plants were cultivated in pots containing a mixture of peat, vermiculite, and perlite (3:2:1, v/v) in a growth chamber. The growth conditions of tea plants were set in 75% relative humidity and 150 μmol/m2/s light intensity with long-day (16 h light at 25°C, 8 h/dark at 18°C). To determine the effects of drought stress on IAA, ABA, GA3, and BR levels and the expression patterns of genes involved in IAA, ABA, GA3, and BR metabolisms, drought stress was simulated by spraying polyethylene glycol (PEG) 6000 (20%, w/v) on tea shoots. The treatment times under drought stress (0, 2, 12, 24, and 48 h) were performed as previously described by Wang et al. (2016). Tea shoots of tea plants under drought stress treatment was collected from each sample, rapidly frozen in liquid nitrogen, and then stored at −80°C for further analysis. Each sample consisted of three biological replicates.
Cytological–anatomical structure analysis
Each fresh sample was cut into small portions (0.5 mm × 0.5 mm) containing a leaf main vein and then stored in formalin acetic acid alcohol solution (ethanol:formalin:acetic acid, 90:5:5, v:v:v) (Servicebio, Wuhan, China). The samples were dehydrated through a graded ethanol series and embedded in paraffin wax. Then, Safranin-O/Fast green was used for histological staining. Subsequently, the samples were mounted in balsam, and then cut into pieces (∼1 mm) using Leica Ultramicrotome (Germany; RM2106). The photograph and imaging system were observed using optical microscope (LEICA DMLS; Leica, Wetzlar, Germany). Optical magnification of microscope was 400 times.
Phytohormone determination
Each sample (500 mg) was ground in an ice-cooled mortar and homogenized with 10 mL of 80% (v/v) methanol extraction solution that contains 1 mM butylated hydroxytoluene for antioxidation. The extract was transferred on a 10 mL centrifuge tube, incubated at 4°C for 4 h and centrifuged at 3500 rpm for 8 min. Supernatant liquid was extracted using Chromoseq C18 solid-phase extraction columns (C18 Sep-Park Cartridge; Waters Corp., Millford, MA). The filtrate was transferred on a 2 mL centrifuge tube and dried through vacuum freeze-drying. Then, 2 mL phosphate-buffered saline that contains 0.1% (v/v) Tween 20 and 0.1% (w/v) gelatin (pH 7.5) was added to make a solution. The extraction and purification of IAA, ABA, GA3, and BR were analyzed via indirect enzyme-linked immunosorbent assay using a previously described method (Yang et al., 2001; Nianjun et al., 2010; Que et al., 2017). In particular, each calibrating sample (50 μL) was placed in 96-well plates with 50 μL immobilized antibody. Then, the solution was washed with washing buffer. Goat anti-rabbit immunoglobulin G (IgG) conjugated with horseradish peroxidase (100 μL) was added in the wells. H2O2 30% (4 μL) was added in the o-phenylenediamine solution and transferred to the wells. H2SO4 2 mol/L (50 μL) was added into each well to terminate the reaction. IAA, ABA, and GA3 were measured at optical density A 490. BR was measured at optical density A 450. The contents of IAA, ABA, GA3, and BR were calculated with the method described by Weiler et al. (1981).
Database search and identification of genes related to IAA, ABA, GA3, and BR metabolisms in tea shoots
The sequences of tea shoots were obtained from the transcriptome data (Wang et al., 2016). The sequences of genes involved in IAA, ABA, GA3, and BR metabolisms were retrieved according to Wang et al. (2015). The nucleotide sequences involved in IAA, ABA, GA3, and BR metabolisms were used as query sequences to identify the related sequences of tea shoots. Alignment of the nucleotide sequences of the IAA, ABA, GA3, and BR metabolisms from transcriptome data of tea shoots was performed by using BioEdit software v.7.0.5.3 (Tom Hall, North Carolina State University, Carolina) (Hall, 1999). The parameter was specified with default value.
Phylogenetic tree construction and sequence analysis
Three web-based tools were used to display phylogenetic tree construction and sequence analysis. Alignment of amino acid sequences was conducted by multiple sequence alignment tool ClustalW. Phylogenetic analyses were constructed using MEGA 6.0 software with neighbor-joining method and visualized on iTOL (Interactive tree of life) by the aligned sequences (Letunic and Bork, 2011). The conserved domains of amino acid sequences were detected on SMART * (Letunic and Bork, 2018). The detail parameters of each web-based tool were set to their default values.
Identification of genes related to IAA, ABA, GA3, and BR metabolisms of tea shoots
The expression profiling analysis of genes involved in IAA, ABA, GA3, and BR metabolisms were calculated through fragments per kilobase of exon per million mapped (FPKM). Gene ontology (GO) enrichment analysis was conducted to identify and classify the enriched genes. The genes involved in different biological pathways were identified through Kyoto Encyclopedia of Genes and Genomes (KEGG) analysis. Expression profiling analysis, GO enrichment, and pathway enrichment analysis were detected using OmicShare tools, a web-based tool for data analysis. The calculated p-value was adjusted through false discovery rate (FDR) correction. The absolute values of FDR ≤0.05 were used as the threshold criteria.
Total RNA isolation, RNA-Seq analysis, and reverse transcription-quantitative polymerase chain reaction analysis
Total RNA was extracted from each sample using an RNA simple Total RNA Kit (Tiangen, Beijing, China) based on the manufacturer's protocol. The concentration of extracted total RNA was measured using a Nanodrop 2000 spectrophotometer (Thermo Scientific, Wilmington, DE). Subsequently, the total RNA of each sample (1.0 μg) was reverse transcribed into complementary DNA (cDNA) using a PrimeScript RT Reagent Kit (TaKaRa, Dalian, China) in accordance with the instructions. cDNA was diluted 16-fold using deionized water for reverse transcription-quantitative polymerase chain reaction (RT-qPCR) analysis.
First, the expression profiles of genes involved in IAA, ABA, GA3, and BR metabolisms were obtained from the RNA-Seq analysis. Then, to confirm the accuracy of the differential expression of IAA-, ABA-, GA3-, and BR-related genes via RNA-Seq in tea shoots, RT-qPCR was performed to on a Bio-Rad BFX96 fluorescence RT-qPCR platform (Bio-Rad, CA). The specific primers were designed by Prime Primer 6.0 software and then synthetized by the GenScript Corporation (Nanjing, China). The reaction program of RT-qPCR was performed as follows: 95°C for 30 s, followed by 39 cycles at 95°C for 10 s, 60°C for 10 s, and 72°C 30 s. Each reaction included 10 μL of Hieff™ qPCR SYBR® Green Master Mix (Yeasen, Shanghai, China), 7.2 μL of deionized water, 2 μL of diluted cDNA strand, 0.4 μL of reverse primer, and 0.4 μL of forward primer. CsGAPDH was used as the reference gene, and the experiments were performed with three independent samples (Wu et al., 2016b).
Statistical analysis
IAA, ABA, GA3, and BR contents, and expression levels of genes involved in IAA, ABA, GA3, and BR metabolisms were analyzed with three independent samples. Experimental data were detected using the Statistical Package for Social Sciences (SPSS) statistics version 17.0 (SPSS, Inc., Chicago, IL). Differences in IAA, ABA, GA3, and BR contents and expression levels of genes involved in IAA, ABA, GA3, and BR metabolisms were performed using Duncan's multiple-range test at a p < 0.05 probability level. Experimental data are presented as the mean ± standard deviation.
Results
Changes in morphology of tea shoots
Tender shoots of tea plants cv. Longjing 43 (included first, second, and third leaf) were used as samples (Fig. 1). Cytological–anatomical structure analysis was performed to detect the effects of drought stress on the morphology of tea shoots. Mesophyll cells were differentiated into spongy tissue (St) and palisade tissue (Pt) cells in the tea shoots. Epidermis (Ep) and mesophyll cells were observed in tea shoots under drought stress (Fig. 2a). Ep, Pt, and St cells were compactly arranged in control group (CK). Pt cells gradually lost their columnar shape with the increase of drought stress time compared with CK.

Photo of tea plant cultivar used in this study.
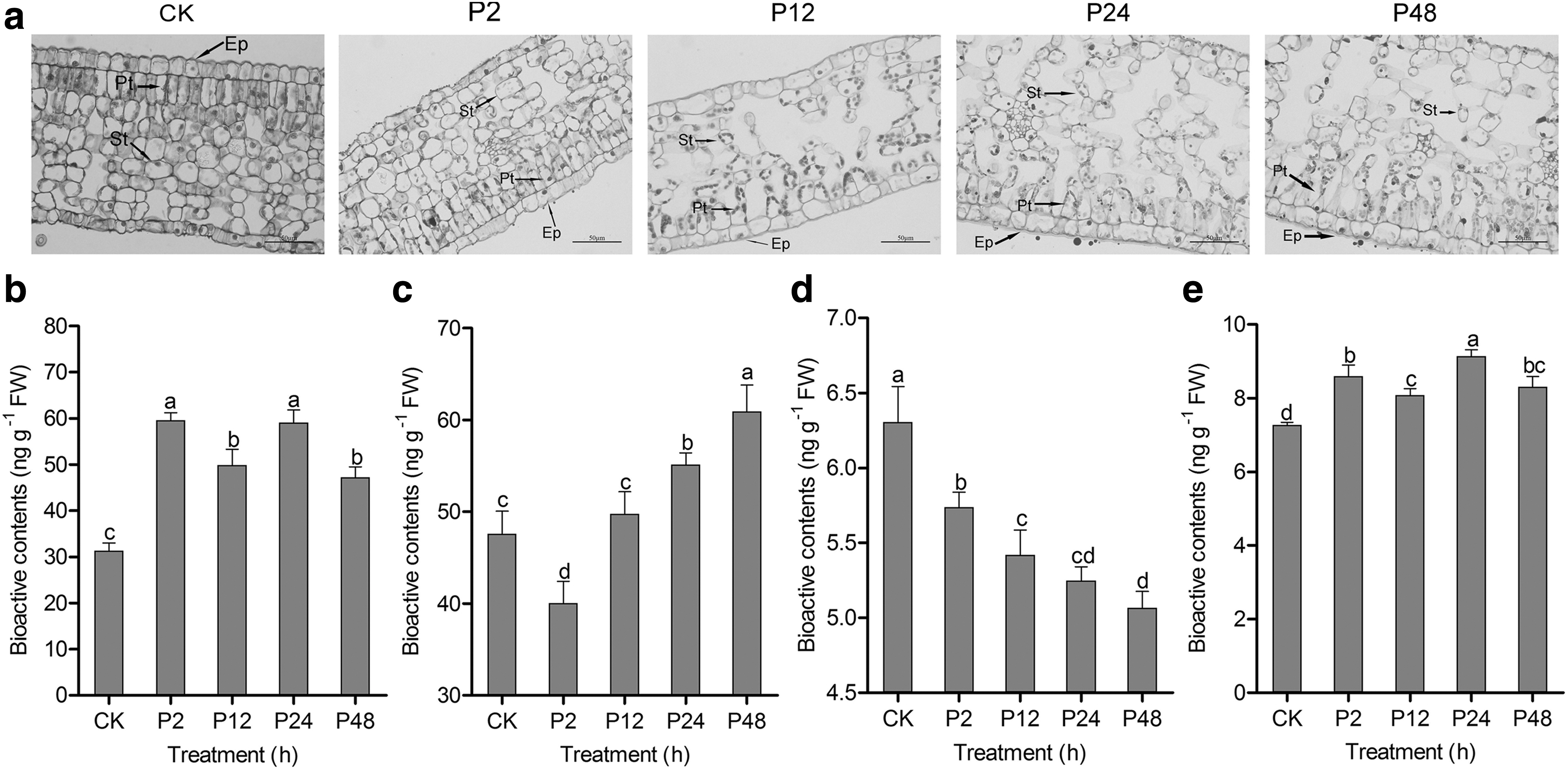
Effects of drought stress on the anatomical structure and dynamic changes of IAA, ABA, GA3, and BR contents in tea shoots.
Dynamic changes in IAA, ABA, GA3, and BR levels
To evaluate whether drought stress could affect the dynamic changes of phytohormone levels in tea shoots, IAA, ABA, GA3, and BR levels were determined (Fig. 2b–e). IAA contents increased at 2 h under drought stress, suggesting that the abundance of IAA might be induced by drought stress. The lowest ABA level was observed at 2 h under drought stress, which gradually increased in the treatment durations ranging from 12 to 48 h. Drought stress significantly decreased the GA3 levels with the increase of treatment time compared with CK. BR levels were increased at 2 h, decreased at 12 h, and peaked at 24 h under drought stress.
Phylogenetic relationships among the genes involved in IAA, ABA, GA3, and BR metabolisms of tea shoots
A total of 60 genes were identified for phylogenetic classification to evaluate the evolutionary relationship among the genes involved in the phytohormone metabolism of tea shoots under drought stress (Fig. 3 and Supplementary Table S1). The nucleotide acid sequences of phytohormone-related genes from tea plant and their putative orthologs from Arabidopsis are given in Supplementary Table S2. Paralogous genes were classified into a gene cluster. For instance, CsGA2ox1 and CsGA2ox1 and CsARF5 and CsARF6 were classified into different gene clusters. The genes involved in IAA and ABA metabolism pathways were classified into the same gene cluster, respectively. For instance, CsTIR and CsNIT2 involved in IAA signal transduction were classified into a gene cluster. CsLUT1 and CsCXHE involved in ABA biosynthesis pathway were classified into a gene cluster. In addition, CsLUT1 and CsCXHE belonged to cytochrome P450 family. This finding suggested that the function of genes might play a role in the evolutionary relationship among the genes involved in biosynthesis, catabolism, response, degradation, and signaling pathways of phytohormones. However, different genes involved in the same metabolism pathway of different phytohormones were classified into same gene cluster. For instance, CsKO and CsCYP81B1, which were the genes involved in GA and IAA biosynthesis pathways, were classified into same gene cluster. CsZDS was the gene involved in ABA biosynthesis pathway. CsDWF7 was the gene involved in BR biosynthesis pathway. CsZDS and CsDWF7 were classified into the same gene cluster. This result indicated that these genes might be involved in regulating the crosstalk between phytohormones.
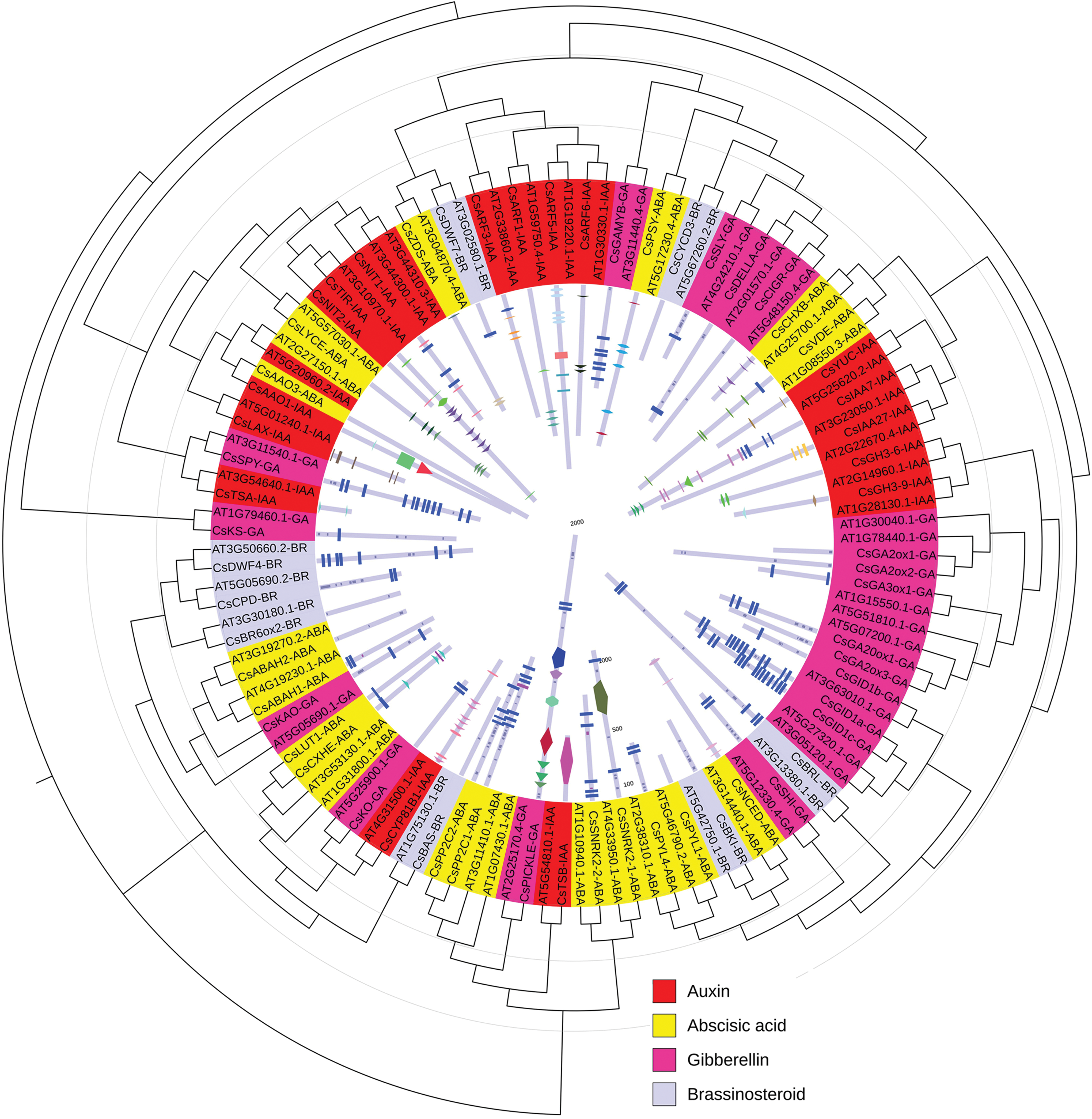
Phylogenetic and structural analysis of the genes and proteins related to IAA, ABA, GA3, and BR metabolisms in tea shoots under drought stress. Color images are available online.
Conserved domains of proteins involved in the IAA, ABA, GA3, and BR metabolisms of tea shoots
The conserved domains of proteins involved in IAA, ABA, GA3, and BR metabolisms under drought stress in tea shoots were identified to determine their characteristics (Fig. 3). The details of these conserved domains in tea shoots and Arabidopsis are listed in Supplementary Table S3. Most of the proteins contained transmembrane and low complexity regions. The number of conserved domains varied from 0 to 17 among the proteins. The largest number of conserved domains was observed in CsPP2C1 protein, which included 10 low-complexity regions and seven transmembrane regions. No conserved domain was observed in CsAAO3 and CsPYL1 proteins. CsAAO1 protein contained CO dehydrogenase flavoprotein C-terminal (CO_deh_flav_C) and aldehyde oxidase and xanthine dehydrogenase, a/b hammerhead (Ald_Xan_dh_C) domains, which existed in the putative protein (AT5G20960.2) in Arabidopsis. Cystathionine beta-synthase domain existed in CsIAA7 protein, which was not observed in the putative protein (AT3G23050.1) of Arabidopsis.
Expression profiles of genes involved in IAA, ABA, GA3, and BR metabolisms in tea shoots based on RNA-Seq data
To elucidate the transcription profiling of genes related to the IAA, ABA, GA3, and BR metabolisms under drought stress in tea shoots, the transcript levels of the genes involved in IAA, ABA, GA3, and BR metabolisms were analyzed based on RNA-Seq data (Fig. 4a–d). The FPKM values are listed in Supplementary Table S4.
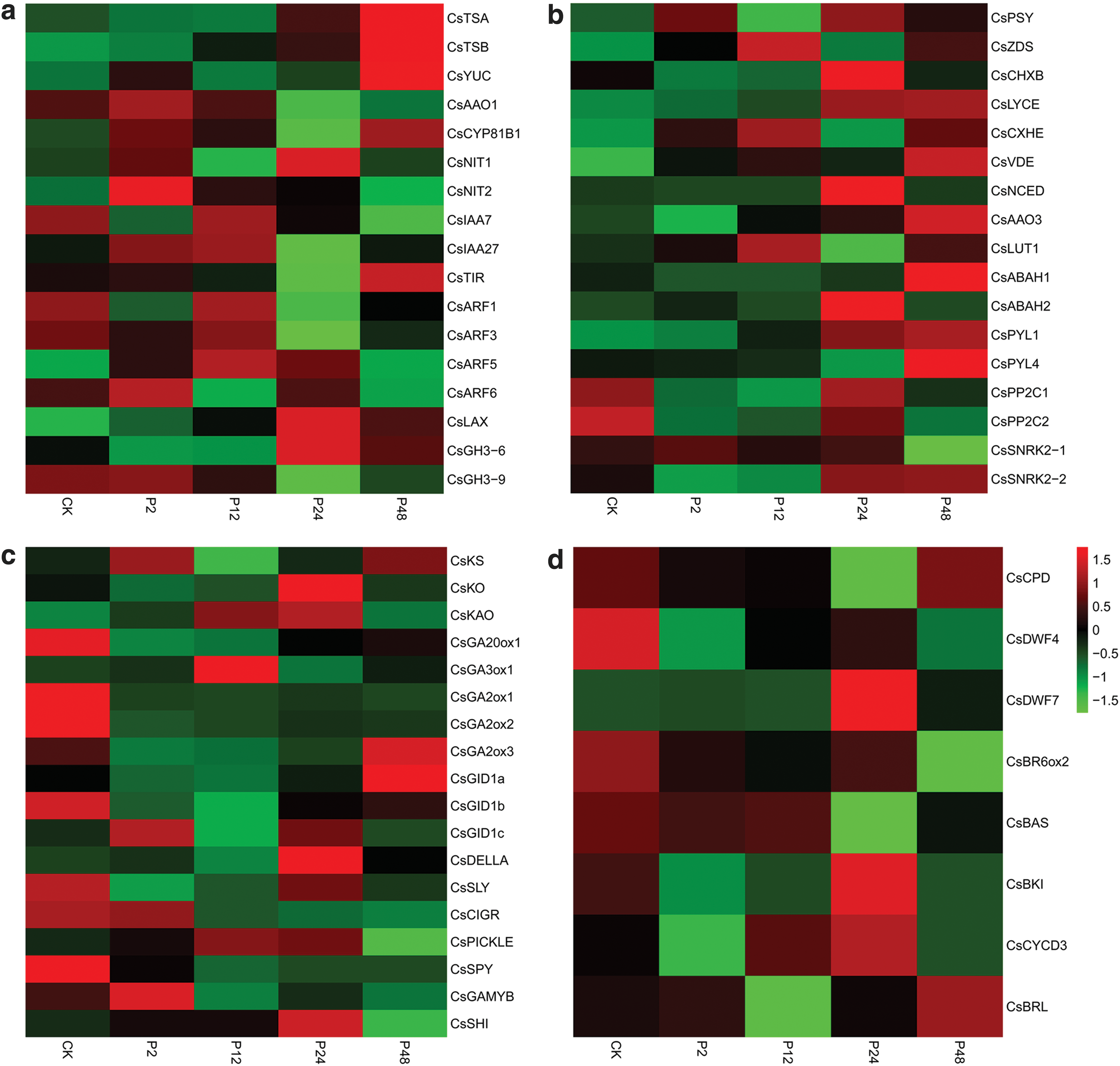
Expression profiling of gene involved in IAA, ABA, GA3, and BR metabolisms in tea shoots under drought stress by RNA-Seq and RT-qPCR analysis.
CsIAA7 expressed a relatively high level at 12 h under drought stress and decreased (Fig. 4a). The transcript level of CsPP2C1 showed a significant increase at 24 h under drought stress (Fig. 4b). The expression level of CsGA2ox3 gradually increased under drought stress. CsSLY reached its peak expression level at 24 h under drought stress and decreased (Fig. 4c). The expression of CsBKI gradually increased, reached its peak at 24 h, and significantly decreased at 48 h under drought stress. CsBAS significantly expressed under drought stress and evidently decreased at 24 h (Fig. 4d).
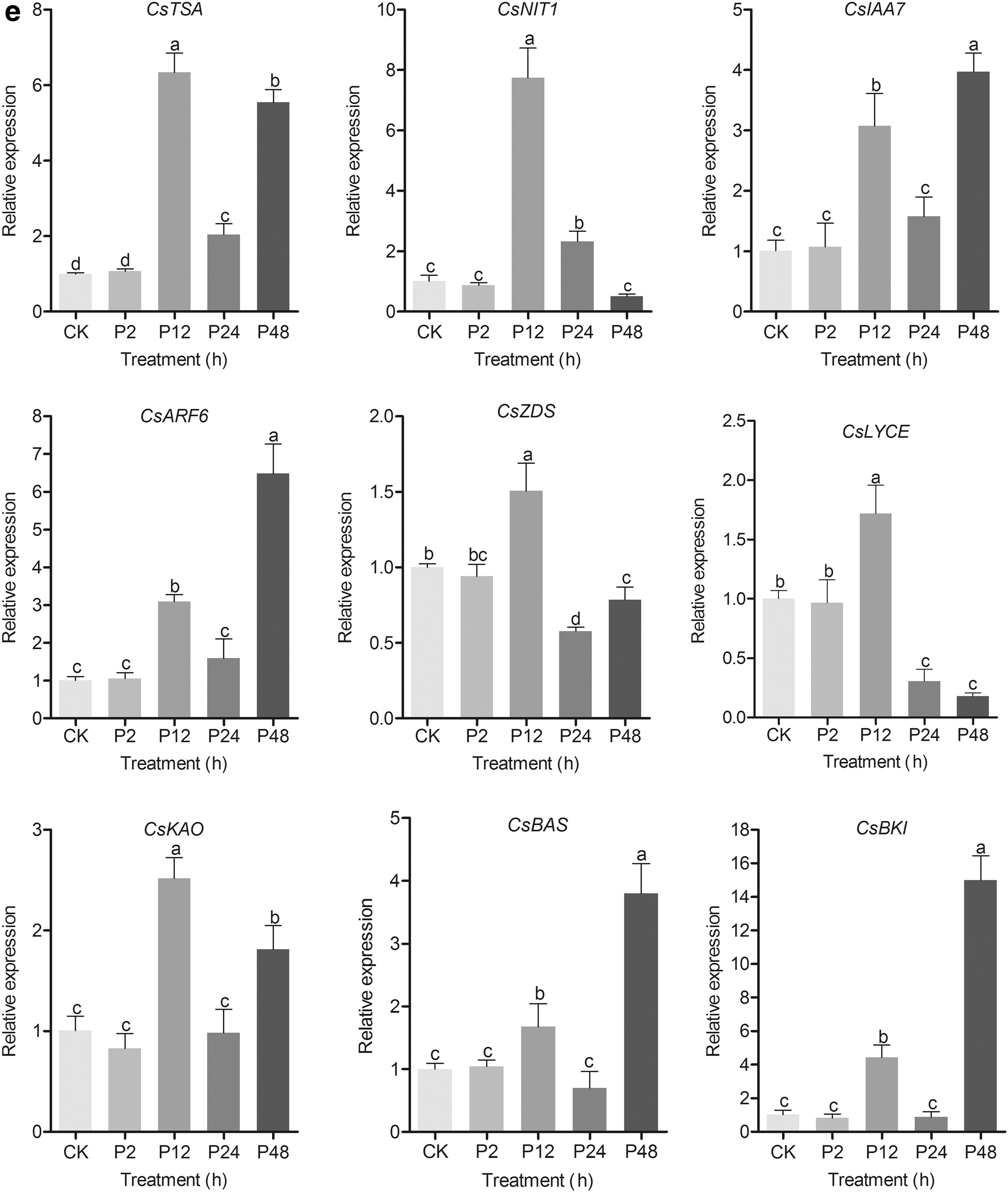
Expression profiles of genes involved in the IAA, ABA, GA3, and BR metabolisms of tea shoot under drought stress based on RT-qPCR analysis
Tea shoots are continuously subjected to various abiotic stresses, such as drought stress, through their lifetime. RNA-Seq analysis showed that a total of nine genes were significantly expressed under drought stress as shown in Figure 4a–d. This finding suggested that they were the crucial genes involved in phytohormone metabolism in response to drought stress. Therefore, the nine genes were analyzed through RT-qPCR (Fig. 4e). The primers of each gene are provided in Supplementary Table S5. The expression level of CsTSA increased at 12 h and decreased at 24 h under drought stress. The expression level of CsIAA7 was similar to the expression level of CsAF6 under drought stress. The expression level of CsNIT1 increased at 12 h and gradually decreased under drought stress. The expression level of CsZDS increased at 12 h under drought stress compared with the control group. The expression level of CsBAS increased at 12 h under drought stress and peaked at 48 h compared with the control group. These results indicated that drought stress could affect the expression profiles of genes involved in IAA, ABA, GA3, and BR metabolisms in tea shoots. The correlations between IAA, ABA, GA3, and BR abundances and transcript abundance of phytohormone-related genes under drought stress are listed in Supplementary Tables S6, S7, S8, S9. A negative correlation was found between CsLYCE expression level and ABA accumulation under drought stress.
Functional classification based on GO annotation
The genes related to IAA, ABA, GA3, and BR metabolisms of tea shoots were classified to determine their assembly, functional annotation, and classification (Supplementary Fig. S1). Cellular process and single-organism process were the most abundant GO slims within the “biological process” category. Metabolic process was a highly represented group in the “biological process” category. Cell and cell part were the most highly represented groups in the “cellular component” category. The number of genes related to binding was less than that in catalytic activity in the “molecular function” category. The detailed results are given in Supplementary Table 10.
Functional classification based on KEGG pathway annotation
The genes involved in IAA, ABA, GA3, and BR metabolisms of tea shoots under drought stress were assigned to different KEGG slims to estimate the interactions and biological functions (Fig. 5). Significant differences in the number of genes were observed in different KEGG pathways. Metabolism and organismal system were the largest group, followed by environmental information processing in various biological pathways. Meanwhile, a total of 13 genes were assigned to signal transduction. The detailed results are provided in Supplementary Table S11.
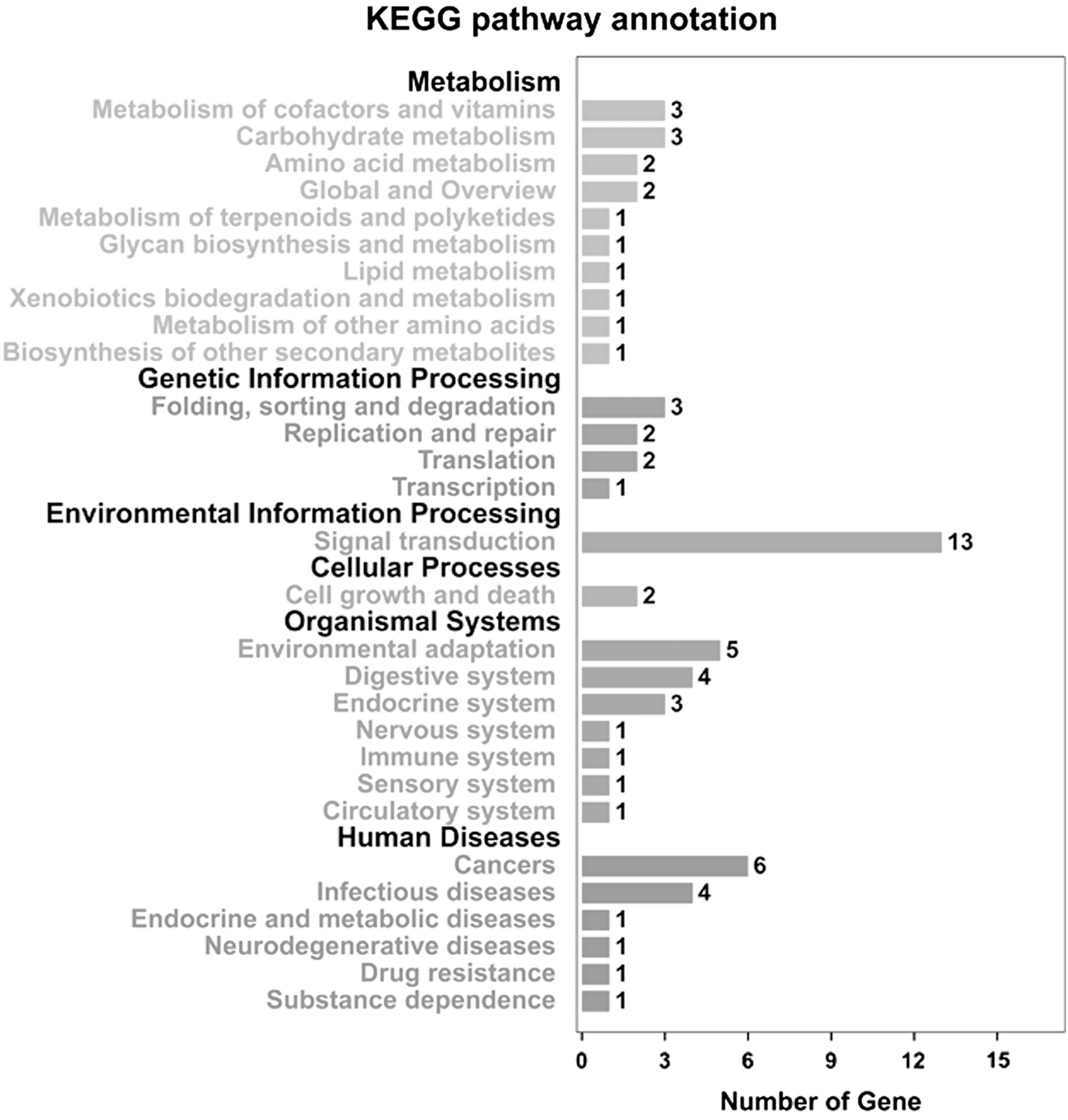
Kyoto Encyclopedia of Genes and Genomes (KEGG) enrichment analysis of genes involved in IAA, ABA, GA3, and BR metabolisms in tea shoots under drought stress.
Distribution of differently expressed genes involved in phytohormone metabolism of tea shoots
The upregulated and downregulated genes were compared to gain considerable insights into the distributions of differently expressed genes related to IAA, ABA, GA3, and BR metabolisms of tea shoots under drought stress (Fig. 6). A total of 10 genes were identified from the comparison analyses of the CK-vs.-P2 library. The number of downregulated genes exceeded that of upregulated genes in the two comparisons (CK-vs.-P2 and CK-vs.-P12). Among the genes, seven downregulated genes and nine upregulated genes were obtained in the comparison between the P12 and P24 libraries. Compared with the P2 library, the number of upregulated genes was the same with that of downregulated genes in the P12 library. Only two downregulated genes were identified between P12 and P48 libraries (P12-vs.-P48). The detailed results are shown in Supplementary Table S12.
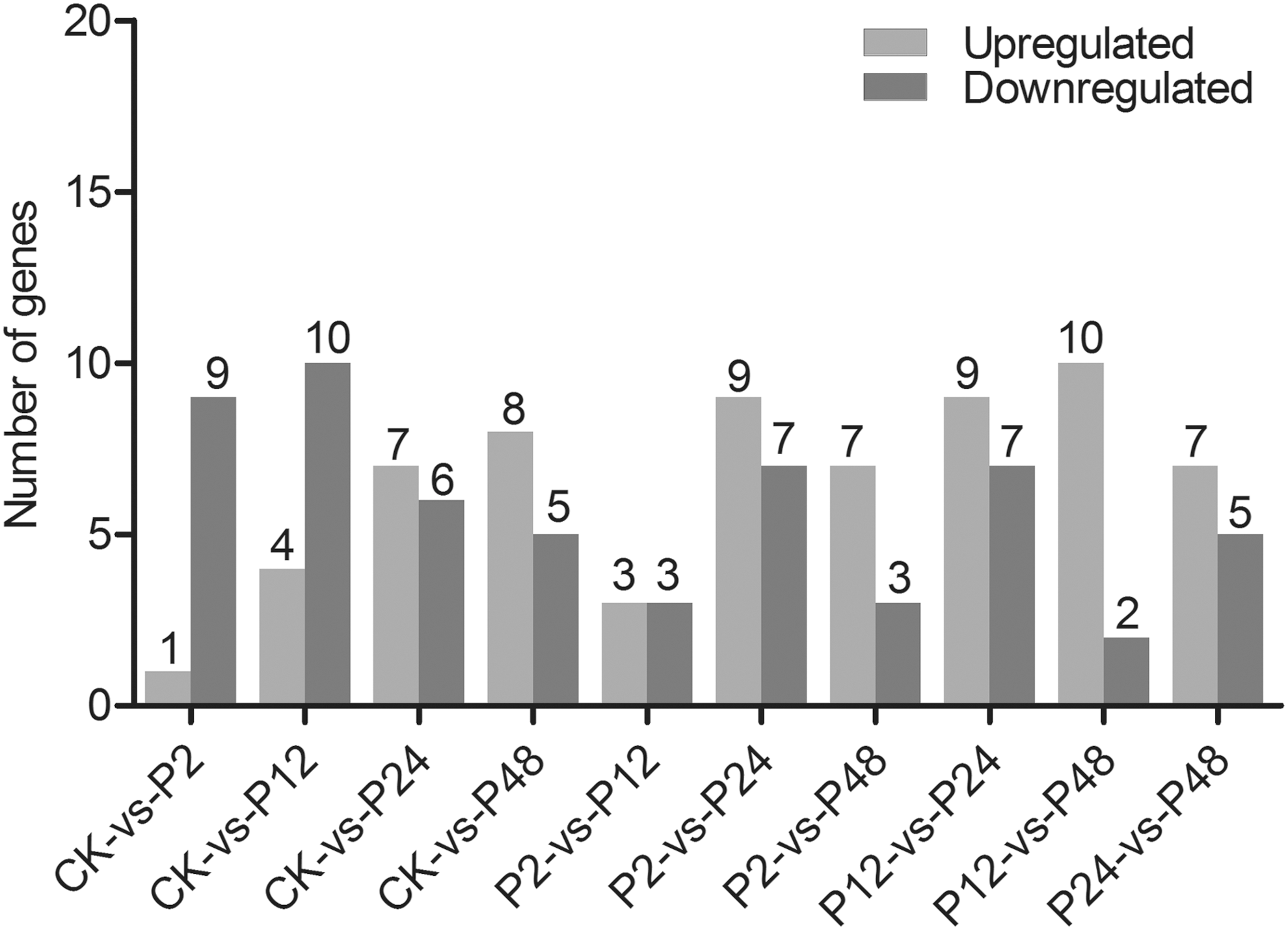
Distribution of genes involved in IAA, ABA, GA3, and BR metabolisms in tea shoots under drought stress. CK-vs.-P2 represents a comparison of upregulated or downregulated genes in CK compared with P2. The labeling is similar for other stages. (CK) Control group. (P2) Drought stress for 2 h. (P12) Drought stress for 12 h. (P24) Drought stress for 24 h. (P48) Drought stress for 48 h.
Discussion
Increasing studies have confirmed that phytohormones could control the transcription and gene expression levels to regulate the cellular division and growth in the plants (Takatsuka and Umeda, 2014). Till now, the genes related to phytohormone dynamic changes of tea shoots under drought stress remain undiscovered.
PEG 6000 is an active polymer and hydrophilic molecule that could cause the dehydration by creating high osmotic pressures in plants, and is mainly used for simulating drought stress. PEG could not pass through the cell wall space of higher plants due to its high molecular weight (Tarkow et al., 1996). Drought stress is related to morphological and plant hormonic changes in tea shoots. Here, to illustrate whether drought stress-induced morphological changes and perturbations exist in hormone metabolism, cytological–anatomical structural analysis was performed. In the research on tea plants, the duration of drought stress of tea plants could be simulated by treating the tea leaves with PEG 6000 treatment. In the present study, we identified the genes related to four phytohormones of tea shoots under drought stress induced by PEG 6000. A total of 60 genes involved in IAA, ABA, GA3, and BR metabolisms were identified from the transcriptome of tea shoots under drought stress.
Characteristics of genes involved in IAA, ABA, GA3, and BR metabolisms of tea shoots
Different genes could be analyzed based on their phylogenetic relationships to determine their classification (Nei, 1996). A total of 60 genes involved in IAA, ABA, GA3, and BR metabolisms were identified to determine their phylogenetic relationships. As illustrated in Figure 3, phylogenetic analysis showed that the paralogous genes were divided into gene clusters. CsGA2ox1 and CsGA2ox1 and CsARF5 and CsARF6 were classified into different gene clusters. Previous studies have reported that all the paralogous genes of TLP (tubby-like protein) genes belong to a subfamily, and these genes might be expanded through gene duplication events in rice and poplar (Yang et al., 2008). Gene duplication plays a vital role in the metabolism of plants (Pichersky and Gang, 2000). Phytohormone metabolism requires the involvement of various related genes in plants (Wu et al., 2016a). Here, we speculated that the paralogous genes under drought stress might have different functions. Gene duplication events might play different roles in the IAA, ABA, GA3, and BR metabolisms of tea shoots.
Difference of expression levels of genes involved in IAA, ABA, GA3, and BR metabolisms of tea shoots
RNA-Seq analysis showed that the expression profiling of CsPYL1 gene involved in ABA signaling pathway was upregulated in tea shoots under drought stress. In Arabidopsis, the expression levels of ABA marker genes were upregulated in response to drought stress (Lim et al., 2013). RT-qPCR analysis showed that the expression profiling of CsIAA7 gene in tea shoots under drought stress increased at 12 h and then decreased at 24 h. The transcript levels of VvAux/IAA4 in Vitis vinifera drought stress decreased by 4 h (Çakir et al., 2013). The expression levels of AaGA2ox1, AaGA2ox2, and AaGA2ox4 in breadfruit were induced by drought stress, whereas salt stress suppressed the expression of AaGA2ox3 (Zhou and Underhill, 2016). The expression levels of CsGA20ox and CsGA2ox were downregulated, and the expression level of CsARF was upregulated at low temperature in tea plants (Pan et al., 2016). Here, RNA-Seq analysis showed that the expression level of CsGA2ox3 in tea shoots under drought stress was upregulated. In summary, our results indicated that the genes involved in phytohormone metabolism of tea shoots under drought stress were differently expressed.
Functional classification, KEGG enrichment, and distribution of genes related to IAA, ABA, GA3, and BR metabolisms of tea shoots
GO could represent the properties of gene products (Gene Ontology Consortium, 2012). Various biological processes could be detected by KEGG pathways analysis (Wrzodek et al., 2013). A total of 60 genes involved in IAA, ABA, GA3, and BR metabolisms were assigned to three ontologies, namely, biological process, cellular component, and molecular function, to determine their functional categories at the molecular level. In the cellular component, the number of genes involved in the membrane was less than that in the cell part. A previous study reported that the cell membranes of Kentucky bluegrass were injured under drought stress conditions (Wang and Huang, 2004). Our finding indicated that the cell and membrane parts were suppressed to prevent the damage induced by drought stress. A previous study showed that the catalytic activity of the phosphatase domain of phytohormone metabolism increased under drought stress (Gong et al., 2003). In this study, we inferred that the catalytic activity of IAA, ABA, GA3, and BR metabolisms played an important role in response to drought stress. Previous studies have revealed that amino acid metabolism and response to abiotic stresses are closely related (Cohen et al., 2010). The analysis of KEGG pathway showed that amino acid metabolism in the IAA, ABA, GA3, and BR metabolism pathways might be suppressed by drought stresses. The number of upregulated genes was more than downregulated genes in the distribution of genes of tea shoots under drought stress. The distribution of upregulated genes was more easily induced than downregulated genes in response to drought stress.
Conclusions
In this work, a total of 60 genes involved in IAA, ABA, GA3, and BR metabolisms were identified. The levels of IAA, ABA, GA3, and BR were significantly changed from 0 to 48 h under drought stress. Our findings suggested that the expression levels of IAA-, ABA-, GA3-, and BR-related genes were significantly induced by drought stress based on RNA-seq and RT-qPCR analyses. Catalytic activity played an important role in the metabolism of IAA, ABA, GA3, and BR under the drought stress. The distribution of upregulated genes was more significantly involved in IAA, ABA, GA3, and BR metabolisms under drought stress than that of downregulated genes.
Authors' Contributions
Conceived and designed the experiments: J.Z., H.L. Performed the experiments: H.L., R.-M.T., J.-X.L., R.-Y.Y., Y.-Z.Y., S.-J.L., M.-H.H., J.-Y.L., and J.Z. Analyzed the data: H.L. Contributed reagents/materials/analysis tools: J.Z. Wrote the article: H.L. Revised the article: J.Z. and H.L. All authors read and approved the final article.
Footnotes
Disclosure Statement
No competing financial interests exist.
Funding Information
The research was supported by the National Natural Science Foundation of China (31570691; 31870681). The funding body did not play any role in study design, data collection and analysis, decision to publish, and preparation of submitting the article.
Supplementary Material
Supplementary Table S1
Supplementary Table S2
Supplementary Table S3
Supplementary Table S4
Supplementary Table S5
Supplementary Table S6
Supplementary Table S7
Supplementary Table S8
Supplementary Table S9
Supplementary Table S10
Supplementary Table S11
Supplementary Table S12
Supplementary Figure S1
References
Supplementary Material
Please find the following supplemental material available below.
For Open Access articles published under a Creative Commons License, all supplemental material carries the same license as the article it is associated with.
For non-Open Access articles published, all supplemental material carries a non-exclusive license, and permission requests for re-use of supplemental material or any part of supplemental material shall be sent directly to the copyright owner as specified in the copyright notice associated with the article.