
Abstract
Silicosis is an occupational disease characterized as inflammatory cells infiltration and severe progressive pulmonary fibrosis. Kaempferol (Kae), a flavonoid that exists in many plants and fruits, has been proved to have anti-inflammatory and antifibrosis functions. However, the effects of Kae on silicosis remain unclear. In the present study, we analyzed the therapeutic effects of Kae in 1-, 7-, and 28-day silicosis models, respectively. In the 1-day model, Kae treatment did not vary the wet-to-dry weight ratios of the lung, apoptotic rate, autophagy, or the expression of inflammatory factors. In contrast, Kae significantly inhibited pulmonary inflammation in the 7-day silicosis models and inhibited silica-induced pulmonary fibrosis in the 28-day models. Besides, we found that Kae partially restored silica-induced LC3 lipidation without increasing the p62 levels. Blocking autophagy with chloroquine antagonized the inhibitory effects of Kae on inflammation, suggesting that autophagy might be required in the therapeutic effects of Kae on silicosis. These findings indicated that Kae inhibits the progression of silica-induced pulmonary fibrosis, which may provide experimental evidences for Kae in the treatment of silicosis.
Introduction
Silicosis is a severe, harmful, and occupational pneumoconiosis disease over the world, especially in China. Inhalation of crystalline silica is the main cause of silicosis. Silicosis exhibits an irreversible progressive fibrosis in lung parenchyma (Fernandez Alvarez et al., 2015; Liu et al., 2019), which causes a heavy burden to the patients both physically and mentally. In recent years, the development of silicosis has been identified as an intricate process involved in the inflammatory response of macrophages, abnormal collagen deposition (Yang et al., 2018), autophagy (Liu et al., 2016, 2017a), and many other biological processes. Because the mechanisms of silicosis are still not completely clear up to now, there are no ideal anti-silicosis drugs at present. Therefore, finding low-toxic, safe, and effective novel drugs for the treatment of silicosis are critical (Fernandez Alvarez et al., 2015).
Kaempferol (Kae) is a natural flavonoid that widely exists in many fruits and vegetables (Kim et al., 2018). Kae is known as an anti-inflammation, antioxidation, and antimicrobes compound, which has been tried in the treatment of many diseases (Liu et al., 2017b), such as cancer, infection, and inflammatory injury (Rajendran et al., 2014; Song et al., 2015; Wilsher et al., 2017; Chitturi et al., 2019). In acute lung injury, Kae exhibits protective effects by modulating TRAF6 polyubiquitination (Qian et al., 2019) and by suppressing oxidative stress, the expression of iNOS and ICAM-1 (Rabha et al., 2018). Recently, Kae is known to alleviate fibrotic airway remodeling through the inactivation of PAR1 in epithelial cell (Gong et al., 2014). Besides, Kae showed significant antifibrosis effects in cardiac remodeling (Liu et al., 2017b). However, whether Kae is able to inhibit silicosis has yet to be understood. The present study is aimed to investigate the anti-silicosis effects of Kae, which may find novel clues for the treatment of silicosis.
Materials and Methods
Chemicals
Silica particles were purchased from Sigma-Aldrich (∼80% between 1 and 5 μm, S5631, St. Louis, MO). Briefly, the silica was baked before use at 180° overnight for sterilization, and then suspended in sterile, lipopolysaccharide-free phosphate-buffered saline (PBS) at a concentration of 40 mg/kg and sonicated for 20 min with an ultrasonic oscillator (Kunshan Instrument Co., Jiangsu, China) to ensure uniform dispersal. Kae was obtained from Sigma-Aldrich and dissolved in corn oil (Sigma-Aldrich) to obtain a 50 mg/mL final concentration solution.
Animals study
All experiments were approved by the Ethics Committee of Peking University People's Hospital (2018PHC069). Six-week-old male C57BL/6J mice were housed under climate-controlled conditions in a 12 h light/12 h dark cycle and accepted free access to food and water. Mice were weighed and randomly allocated to each group. The silicosis models were constructed according to previous description (Jessop et al., 2016). In brief, silica was dried and suspended in sterile PBS. Particulates were sonicated for 20 min before intratracheal instillation. For Kae treatment, the mice were intraperitoneally injected with 150 mg/kg Kae in corn oil daily. Equal volume of vehicle was administrated intraperitoneally in both of the control and silicosis groups. For chloroquine (CQ) treatment, mice were intraperitoneally injected with 50 mg/kg of CQ (Sigma-Aldrich) in sterile PBS.
Wet-to-dry weight ratio measurement
To investigate whether Kae could inhibit silica-induced pulmonary edema, silica crystalline suspension was administered via intratracheal injection (0.15 g/kg). Twenty-four hours after the injection, the whole lungs in each mouse were obtained for the wet-to-dry weight ratio analysis. For Kae treatment, the mice were pretreated with Kae for 24 h before intratracheal injection and a second injection of Kae after silica treatment. The lung tissues were excised and weighed immediately to obtain the wet weight. After being baked at 68°C for 48 h, the tissues were weighed and obtained dry weight. The calculation index was performed according to the following formulas: lung wet-to-dry weight ratio = weight of the whole wet lung/weight of the whole dry lung.
Real-time qPCR
Total RNA was isolated from lung tissues using the TRIzol Reagent (Invitrogen, Carlsbad, CA) and reverse transcribed into cDNA with PrimeScript RT kit (Takara, Japan). Real-time quantitative PCR (qPCR) was performed using TB Green® Premix Ex Taq™ II (Takara) for the evaluation of expression levels of IL-1β, IL-4, IL-6, IL-13, MCP-1, TIMP-1, TGF-β1, and TNF-α. All of the primer sequences are listed in the Supplementary Table S1. GAPDH was used as an internal control, and the expression of each gene was normalized using 2−ΔΔCT assays.
Histological analysis
The lung tissues were fixed in 10% formalin, embedded in paraffin, and cut into 5 μm thick slices. The slices were de-waxed and routinely stained with hematoxylin and eosin for histological analysis. Masson's trichrome staining was performed to analyze the degree of pulmonary fibrosis. The fibrotic area (FA) and total area (TA) were measured using Image-Pro Plus 6.0 (IPP 6.0) software. The fibrotic degree was evaluated using the FA/TA ratio. For analyzing apoptosis, TUNEL staining was performed using the ApopTag Peroxidase In situ Apoptosis Detection Kit (Millipore, Billerica, MA) according to the manufacturer's protocol. The TUNEL+ cells were counted in five random fields in each slide, and the TUNEL+ cells/total cells ratios were calculated for indicating the apoptosis.
Western blot analysis
Total protein from tissue or cellular samples was extracted with RIPA lysis buffer, quantified, and normalized. Equal amounts of proteins were loaded and separated using SDS-PAGE. The proteins were blotted onto a nitrocellulose membrane and blocked with 5% nonfat milk at room temperature for 1 h followed by the incubation with the following primary antibodies at 4°C overnight: rabbit anti-LC3 (Sigma-Aldrich), rabbit anti-p62, rabbit anti-Beclin-1 (Santa Cruz, CA), rabbit anti-p70S6K, rabbit anti-p-p70S6K, rabbit anti-GAPDH (Cell Signaling Technology, MA), and mouse anti-β actin (Sigma-Aldrich). Then, the membranes were washed three times with TBST and incubated with HRP-conjugated goat anti-rabbit or mouse secondary antibodies (ZSGB-BIO, Beijing, China) for 1 h at room temperature. Subsequently, the specific bands were visualized and analyzed using Quantity One software V 4.6.2 (Bio-Rad, Hercules, CA).
Gelatin zymography
For the measurement of matrix-metalloproteinase (MMP)-2 and MMP-9 activities, gelatin zymography was performed as previously described. Briefly, lung tissues were lysed with RIPA, quantified, and diluted with 2 × native loading buffer. Equal proteins were immediately loaded onto an SDS-PAGE containing 0.08% of gelatin. The proteins were separated and washed three times with 2.5% Triton X-100. Then, the gel was immersed in a reaction buffer (containing 50 mM of Tris [pH 7.5], 150 mM of NaCl, and 10 mM CaCl2) and incubated at 37°C for 16 h. The gel was then stained with Coomassie Brilliant Blue and visualized using Quantity One software V 4.6.2 (Bio-Rad).
Statistical analysis
Statistical analyses were performed using SPSS 19.0 software (SPSS, Inc., Chicago, IL). Two-tailed Student's t-test or one-way ANOVA was used for analyzing differences. Data are expressed as the mean ± standard error of the mean and p < 0.05 was considered statistically significant.
Results
Kae do not vary silica-induced acute lung injury
The inhalation of silica crystalline causes acute inflammation, and thus induces pulmonary edema (Sharawy et al., 2013). We found that the lung wet-to-dry weight ratio in silica-treated mice were indeed elevated compared with the mice in control group (Fig. 1). However, this ratio remained comparable between the mice of the silica-treated and the silica+Kae-treated group (Fig. 1A). Apoptosis plays an important role in the development of acute lung injury (Shao et al., 2017). Therefore, we perform TUNEL staining to analyze the change of apoptosis in silica-induced acute lung injury. We observed that silica significantly induced apoptosis in the lung (Fig. 1B, C). However, Kae failed to restore the apoptotic rate induced by silica installation (Fig. 1B, C). We also detected the changes of autophagic markers in the lung. We found that silica treatment slightly inhibited the LC3 lipidation in the lungs of 1-day silicosis models, whereas Kae did not show significant effects on autophagy markers including LC3, Beclin-1, or p62 expression (Fig. 1D).To further investigate inflammatory changes in the lungs, we detected the expression of IL-1β, IL-4, IL-6, IL-13, MCP-1, TIMP-1, TGF-β1, and TNF-α, which are key inflammatory factors in the lung injury. We observed that the expression of IL-1β, IL-6, MCP-1, TIMP-1, and TGF-β1 were significantly increased, whereas the expression of IL-4 and IL-13 were decreased in the lung of silica-treated mice (Fig. 2). However, the administration of Kae did not vary the expression of these inflammatory factors (Fig. 2), suggesting that Kae might not inhibit silica-induced acute lung injury.

Kae fails to inhibit silica-induced acute lung injury. Sterile silica suspension was intratracheally injected and vehicle injection was used as controls. Kae was injected intraperitoneally. Tissues were harvested at 1 day after the treatment.
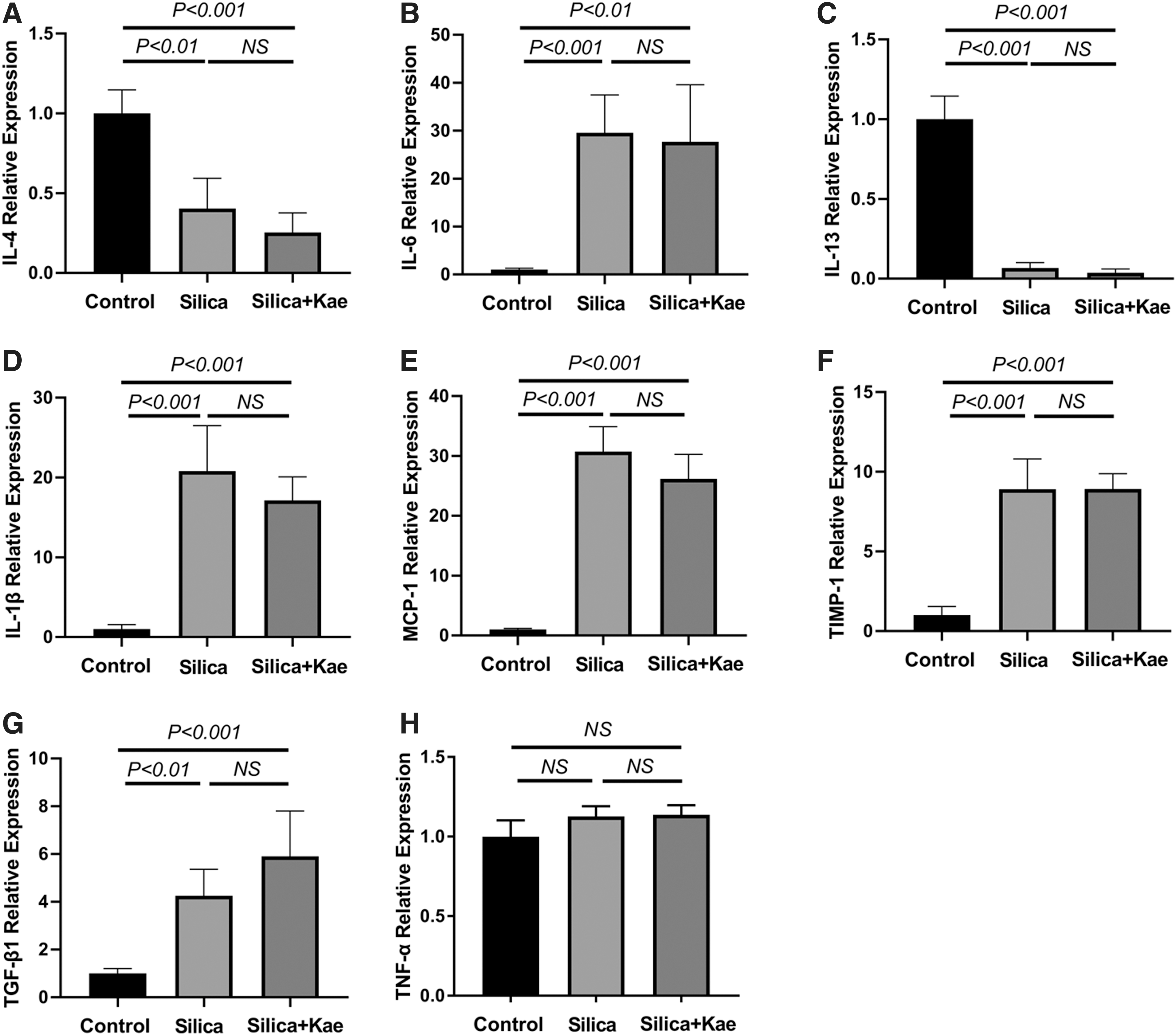
Kae did not vary pulmonary inflammation in 1-day silicosis models.
Kae inhibits silicosis in the 7-day models
It has been reported that early silicotic nodules are composed of accumulated macrophages (Zhang et al., 2014; Chen et al., 2015; Shichino et al., 2015). To investigate the effects of Kae on silicosis, we constructed a 7-day silicosis mice model. After 7 days of the intratracheal injection of silica crystalline, the silicotic nodules were formed with accumulated cells (Fig. 3A). The size of silicotic nodules appeared to be smaller in the presence of Kae (Fig. 3A). The production of MMPs, such as MMP-2 and MMP-9 lead to breakdown of extracellular matrix (ECM) and influence the development of silicosis (Langley et al., 2011; Li et al., 2013). In the 7-day silicosis model, we found that the activities of both MMP-2 and MMP-9 were both upregulated by silica treatment, and these effects could be attenuated by Kae treatment (Fig. 3B).
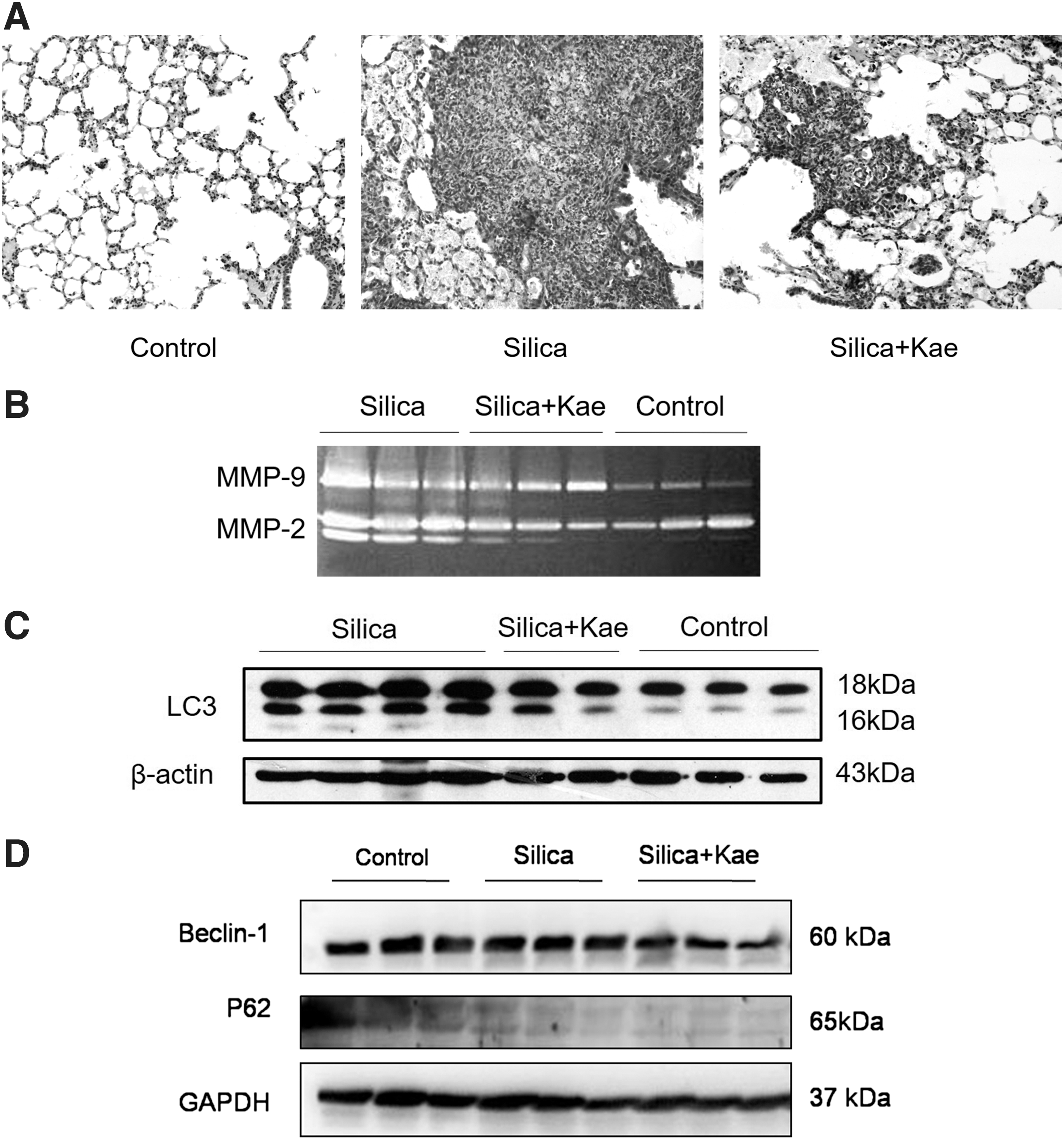
The effects of Kae on the 7-day silicosis models.
Autophagic flux is required in the inhibitory effects of Kae on silica-induced inflammation
Autophagy, as a classical cell death pathway, plays an important role in pulmonary fibrosis (Chen et al., 2015). Upon exposing with silica, macrophages engulf the dust particles and then removed them from the lungs. Because the macrophages cannot dissolve the silica particles, autophagy could be constantly activated and thus trigger a series of inflammatory reactions (Liu et al., 2016). In addition, the increased autophagic activity has been associated with the cumulative level of silica dust exposure to the lungs, triggering the activation and apoptosis of macrophages, ultimately inducing cell death and the development of fibrosis (Chen et al., 2015; Liu et al., 2017a). Therefore, we analyzed the status of autophagy in the 7-day silicosis model. We found that silica induced a significant activation of autophagy in the lung tissues, whereas combined treatment with Kae partially antagonized the autophagy activation (Fig. 3C). However, Kae treatment did not upregulate the p62 levels, indicating that autophagic flux may not be inhibited (Fig. 3D). Consistently, the levels of Beclin-1 could not be varied by Kae in the 7-day silicosis models (Fig. 3D). These data indicated that Kae may antagonize silica-induced autophagy without inhibiting autophagic flux in the early phase of silicosis.
To investigate the role of autophagy in silicosis, we blocked autophagic flux with CQ. This drug is known to have anti-silicosis effects (Allison et al., 1966; Zhao et al., 2019). Consistently, our data indicated that CQ restored the levels of most observed inflammatory factors compared with the silicosis group (Fig. 4). Kae showed more significant effects on the inhibition of the inflammatory factors (Fig. 4). More interestingly, CQ could block the inhibitory effects of Kae on the expression of IL-4, IL-6, IL-1β, MCP-1, TIMP-1, TGF-β1, and TNF-α (Fig. 4). These data indicated that autophagic flux was required in Kae's anti-silicosis effects.

Autophagic flux is required in the anti-inflammatory effects of Kae on silicosis.
Kae attenuates silica-induced pulmonary fibrosis in mice
Silicosis is characterized as progressive pulmonary fibrosis. To explore the effects of Kae on silica-induced pulmonary fibrosis, a 28-day silica exposure mouse model was constructed. Data from Masson's trichrome staining indicated that silica exposure increased the collagen deposition. In contrast, combination treatment with Kae attenuated the collagen deposition in the lung (Fig. 5A, B). Besides, we observed that both MMP-2 and MMP-9 were activated in the 28-day silicosis models (Fig. 5C) and the activity of MMP-2 could be inhibited by Kae (Fig. 5C). However, Kae failed to inhibit the MMP-9 in the 28-day silicosis mice (Fig. 5C). These data together indicated that Kae was able to attenuate silica-induced pulmonary fibrosis and inhibited MMP-2 activation.

Kae inhibits silica-induced pulmonary fibrosis.
We next analyzed the effects of Kae on autophagy in the 28-day silicosis model. Silica exposure induced a significant increase of LC3-II in the lung tissues, which indicated an activation of autophagy. However, Kae failed to restore autophagy levels in this model (Fig. 5D). We then analyzed the phosphorylation status of p70S6K (T389) as a known substrate of mTOR, which is widely used as an indicator of mTOR activation (Lee et al., 2016; Takegaki et al., 2017). mTOR is a pathway that is closely associated with protein synthesis (von Walden et al., 2016). We found that silica exposure induced a significant reduce of phosphorylated p70S6K at the site of T389, and Kae augmented the phosphorylation of p70S6K, indicating that the mTOR signaling pathway was inhibited by Kae (Fig. 5D). The inhibition of mTOR caused by Kae treatment may reduce protein synthesis and thus contribute to the inhibition of collagen deposition.
Discussion
Silicosis is a fatal and irreversible occupational disease with limited treatment options (Lakatos et al., 2006). Although it has taken many security measures for the protection of miners, there are still many silicosis patients all over the world. Therefore, finding novel therapy and drugs is urgent for this disease. In this study, we found that Kae, a compound of flavonols, might attenuate the progression of silicosis in our experimental models. Kae alleviated silica-induced inflammation, collagen deposition, and modulate the autophagy activity. These findings provided indicated that Kae might be a potential therapeutic drug for silicosis.
Recent studies have reported that Kae had the function of anti-inflammation, antioxidation, and anticancer (Rajendran et al., 2014; Devi et al., 2015). Kae suppressed the activation of the HMGB1/TLR4 inflammatory pathway in striatum tissues of mice (Yang et al., 2019). In addition, Kae reduced oxidative stress by inhibiting TBARS and 3-NT formation through downregulation of CYP2E1 expression and upregulation of UGT1A1 expression (Tsai et al., 2018). The anticancer effect of Kae in pancreatic cancer was mediated by inhibition of EGFR related Src, ERK1/2, and AKT pathways (Lee and Kim, 2016). What is more, it has been confirmed that Kae prevented and reversed ventricular airway and fibrosis via bronchial EMT by modulating PAR1 activation (Gong et al., 2014), suggesting Kae have a potential inhibiting effect on the development of fibrosis. Our data revealed that Kae attenuated silica-induced pulmonary fibrosis and inflammation in the lung tissues, which indicated that Kae had an inhibiting effect on silicosis.
The pathophysiology of silicosis includes deposition of silica particles into the lung alveoli. These particles are absorbed by alveolar macrophages, irritating an inflammatory response, which stimulates the proliferation of fibroblasts and production of collagen. Silica particles are wrapped by collagen, resulting in fibrosis and the nodular lesions and eventually the formation of silicosis (Pollard, 2016). However, insufficient research about the pathological mechanisms of collagen accumulation and remodeling in silicosis has severely restricted of effective therapeutic strategies (Beamer et al., 2010). It has been proved that increased MMP-2 and MMP-9 promote ECM degrading and remodeling, which causes damage to the basal lamina of alveolar epithelium and the invasion of fibroblasts into alveolar cavity (Langley et al., 2011; Li et al., 2013). In the present study, we observed that MMP-2 and MMP-9 activity decreased at 7 and 28 days Kae treatment, which indicated that Kae reduced gelatinase in silica-induced pulmonary fibrosis.
Inflammation in the early phase of silicosis is a key process for the initiation of pulmonary fibrosis in the late phase. We found that Kae significantly suppressed inflammation in the 7-day models, and these effects might be associated with autophagy. Autophagy is a universal process that is closely linked with the degradation of protein aggregates and organelles. Previous studies indicate that autophagy in the macrophages can be activated by silica, characterized as the accumulation of autophagosomes, which may be associated with the silicosis progression (Chen et al., 2015; Liu et al., 2016, 2017a). However, the activation of autophagy also attended in suppressing pulmonary fibrosis and inflammation in silicosis (Han et al., 2016; Jessop et al., 2016). Therefore, the function of autophagy in silicosis remains unclear and may play different roles according to the different stages of silicosis. Although we found that Kae could inhibit silica-induced LC3 lipidation in the 7-day silicosis model, the levels of p62, a biomarker for autophagic flux, was not increased, indicating that Kae might not inhibit autophagic flux in the silicosis models. More interestingly, blocking autophagic flux with CQ antagonized the anti-inflammatory effects of Kae, suggesting that autophagic flux was required.
The mTOR signaling pathway is a classical autophagy pathway (Serrano-Oviedo et al., 2018), which is known to directly regulate protein synthesis (Kim and Guan, 2015). Our data show that Kae treatment induced a significant inhibition of mTOR in the 28-day silicosis models, which may downregulate the protein synthesis in the lung. The histological analysis indicated that collagen deposition is a predominant pathological change in this stage of silicosis. Therefore, we postulate that the inhibition of mTOR induced by Kae may help to reduce the synthesis of collagen and thus attenuate the progression of pulmonary fibrosis.
In conclusion, our data demonstrated that Kae alleviates silica-induced pulmonary fibrosis and inflammation associated with the regulation of autophagy and attenuated gelatinase, which suggests that Kae may be a candidate compound for the treatment of silicosis. The further study will focus on the detailed mechanism of Kae on autophagic flux in silicosis.
Footnotes
Disclosure Statement
The authors declare no conflicts of interest.
Funding Information
This work was supported by the Capital Clinical Application Research (Z181100001718156) and Special Financial Grant from the China Postdoctoral Science Foundation (2017T100052).
Supplementary Material
Supplementary Table S1
References
Supplementary Material
Please find the following supplemental material available below.
For Open Access articles published under a Creative Commons License, all supplemental material carries the same license as the article it is associated with.
For non-Open Access articles published, all supplemental material carries a non-exclusive license, and permission requests for re-use of supplemental material or any part of supplemental material shall be sent directly to the copyright owner as specified in the copyright notice associated with the article.