
Abstract
Although advances have been made in the development of antiangiogenesis targeted therapy and surgery, metastatic clear cell renal cell carcinoma (ccRCC) is still incurable. Activation-induced cytidine deaminase (AID) is mainly expressed in a variety of germ and somatic cells, and induces somatic hypermutation and class-switch recombination, playing a vital role in antibody diversification. We confirmed that AID was expressed at a higher level in ccRCC tissues than in the corresponding nontumor renal tissues. We explored the impact of AID on ccRCC proliferation, invasion, and migration. In 769-p and 786-0 cells, expression of an AID-specific short hairpin RNA significantly reduced AID expression, which markedly inhibited tumor cell invasion, proliferation, and migration. Previous studies showed that AID is associated with Wnt ligand secretion mediator (WLS/GPR177), cyclin-dependent kinase 4 (CDK4), and stromal cell-derived factor-1 (SDF-1/CXCL12) regulation, which was further confirmed in human ccRCC tissues. Therefore, we studied the relationship between AID and these three molecules, and the impact of AID on epithelial-to-mesenchymal transition in ccRCC. WLS/GPR177, SDF-1/CXCL12, and CDK4 were sensitive to 5-azacytidine (a DNA demethylation agent), which reverted the inhibition of carcinogenesis caused by AID repression. In summary, AID is an oncogene that might induce tumorigenesis through DNA demethylation. Targeting AID may represent a novel therapeutic approach to treat metastatic ccRCC.
Introduction
Among renal cancers, renal cell carcinoma (RCC) is the most common solid tumor, whose occurrence has increased gradually by 2–4% annually over the last few decades (Zhou et al., 2019). Clear cell renal cell carcinoma (ccRCC) is the most common pathological subtype of RCC, accounting for ∼90% of all RCCs, and overall mortality rates for ccRCC also show an upward trend (Ljungberg et al., 2019). In patients with ccRCC, surgery and targeted therapy are the most used therapies because chemotherapy and radiotherapy are not particularly effective (Zhang et al., 2018). In recent years, significant advances have been made in terms of several targeted therapy factors, including AXL receptor tyrosine kinase, receptor tyrosine kinase (Stone, 2015), mammalian target of rapamycin (Ghidini et al., 2017), and vascular endothelial growth factor (Sharpe et al., 2013) in the clinical treatment of ccRCC. However, after partial or radical nephrectomy, a subsection of patients with localized ccRCCs develop metastases, which limits the efficacy of targeted therapies (Motzer, 2016; Zhang et al., 2018). Therefore, further novel markers that participate in metastasis and tumorigenesis of ccRCC are urgently required for use as potential therapeutic targets.
Activation-induced cytidine deaminase (AID) is a member of the AID/apolipoprotein B mRNA editing complex catalytic polypeptide family (Conticello, 2008; Munoz et al., 2013), which causes cytosine deamination to uracil, followed by repair through base excision repair or mismatch repair. These are error-prone processes that cause mutations leading to dU:dG mismatches (Bhutani et al., 2011). AID, through its cytidine deaminase function, is a key enzyme that initiates somatic hypermutation, class-switch recombination (CSR), and gene conversion, which regulate the diversification of the Ig genes in activated germinal center B lymphocytes (Matsumoto et al., 2010; Zan and Casali, 2013). Within repetitive heterochromatin, an increased frequency of recombination events and retrotransposon activation are the result of demethylation, leading to genomic instability (Andersen and Jones, 2013). Some studies indicated that AID participates in the demethylation of methylcytosine in mammalian DNA (Morgan et al., 2004; Rai et al., 2008; Bhutani et al., 2010). DNA methylation is a major epigenetic mechanism of gene repression (Cortellino et al., 2011) and in mammals, it is essential for embryonic development, stem cell pluripotency (Youn et al., 2018), and somatic tissue differentiation.
The regulation of AID is complicated in germinal center B cells. AID is regulated by multiple factors, such as NF-κB, homeobox C4, paired box 5, transmembrane activator and CAML interactor, toll-like receptors, CD40 molecule and SMAD family member 3/4 (Zarnegar et al., 2004; Pone et al., 2012; Xu et al., 2012). In addition, it enhances AID expression, as induced by stimuli inducing interleukin-4, transforming growth factor-β, tumor necrosis factor-α (Duan et al., 2016), and CSR. AID expression must be regulated to avoid damage caused by its dysregulation, for example, chromosomal translocations, and to sustain genomic integrity (Robbiani et al., 2008, 2009; Hasham et al., 2010). This tight regulation is effected through fine control of AICDA transcription (the gene encoding AID) (Stavnezer, 2011), post-transcriptional and post-translational regulation (especially AID's nuclear and cytoplasmic distribution) protein stability (Hasler et al., 2011), and activity regulation (Li et al., 2012). AID dysregulation and misexpression not only cause DNA damage to Ig genes but also to a variety of other genes, contributing to inflammatory diseases and tumorigenesis (Okazaki et al., 2003; Staszewski et al., 2011). In certain human malignancies, such as breast cancer (Munoz et al., 2013), leukemia (Wright et al., 2015), gastric adenocarcinoma (Mohri et al., 2017), and colon cancer (Araki et al., 2019), AID is a crucial molecule that promotes the metastasis and proliferation of cancer cells.
Epithelial-to-mesenchymal transition (EMT) is the driving force behind normal morphogenesis and tumor metastasis. Evidence has shown that EMT requires AID for the development process from nonmalignant mammary epithelial cells to relative chronic inflammatory cells to malignant tumors. Meanwhile, during this process, the expression level of AID constantly increases (Munoz et al., 2013). Increased AID expression results in loss of the adhesion molecule biomarker E-cadherin and upregulated expression of Vimentin or N-cadherin during EMT, together with abnormal cell polarity and accelerated tumor proliferation and metastasis (Montamat-Sicotte et al., 2015). In addition, DNA demethylation through AID might influence the expression of tumor progression factors (Li et al., 2019). Thus, AID regulates gene expression in complex developmental processes. Dysregulation of AID expression is closely related to the occurrence and progression of tumors. However, its precise role and mechanism in the metastasis and proliferation of ccRCC remain poorly understood.
In a previous study, we found that AID is associated with Wnt ligand secretion mediator (WLS/GPR177), cyclin-dependent kinase 4 (CDK4), stromal cell-derived factor-1 (SDF1/CXCL12), and β-catenin regulation in ccRCC (Li et al., 2019). In this study, we analyzed AID protein levels in ccRCC tissues and their corresponding nontumor tissues. Meanwhile, a model of AID knockdown in 786-0 and 769-P cells was established to determine whether AID can induce the expression of WLS, CDK4, and SDF1 through demethylation in the progression of ccRCC. In vitro, we studied the impact of altered AID expression on 786-0 and 769-P cell proliferation and metastasis. AID significantly promoted the malignant phenotype of ccRCC and sensitized ccRCC cells to proliferation and metastasis, possibly through regulation of downstream genes WLS, CDK4, and SDF1. Furthermore, through DNA demethylation, AID might enhance ccRCC invasiveness and affect the EMT of ccRCC cells. Collectively, the results of this study provide an explanation for the aggressiveness of ccRCC.
Materials and Methods
Patient selection and preparation of tissue
We enrolled 36 patients diagnosed with ccRCC from March 24, 2017 to May 19, 2018 at Renmin Hospital of Wuhan University (approval no. 2017K-C015). An experienced pathologist identified the renal cancer tissue specimens and their corresponding nontumor renal tissues. The samples were stored in liquid nitrogen or 4% formalin fixed before the Western blotting, immunohistochemistry (IHC), hematoxylin and eosin (HE) staining, or immunofluorescence experiments. All experiments were carried out according to the Code of Ethics of the World Medical Association, and the ethics committee of Renmin Hospital of Wuhan University authorized the study. All the human subjects provided informed consent for the use of their samples and data.
Cell lines and their culture
In this study, we purchased human ccRCC cell lines (769-P, 786-0, A498, and ACHN) and human renal proximal tubule epithelial (HK2) cells from ASY Biotechnology Ltd., Corp. (Wuhan, China). All cells were grown in 89% Roswell Park Memorial Institute (RPMI) 1640 Medium (cat. no. C11875500BT; Life Technologies, Gibco, Carlsbad, CA) with 10% FBS (cat. no. C10010500; Life Technologies) and 1% penicillin–streptomycin (Gibco). The cells were grown at 37°C with 5% carbon dioxide (CO2) in a humidified incubator. Trypsin digestion was carried out on 80–90% confluent cells from shake flask cultures, after which the cells were collected for subsequent experiments.
Construction of the lentivirus vector expressing an AID-specific short hairpin RNA and cell transfection
Two short hairpin RNA (shRNA) oligonucleotide sequences targeting the human AICDA gene were designed. The negative control comprised of a nonspecific scrambled shRNA sequence. The shRNA sequences comprised of shRNA1 (siRNA1, 5′-TGACTTACGAGACGCATTT-3′) and shRNA2 (siRNA2, 5′-TTTCGTACTTTGGGACTTT-3′). These sequences were synthesized by GeneChem Corp. (Shanghai, China) and labeled with the GFP. We then ligated the AID-specific shRNAs (shAICDA) and the nonspecific sequence (shCtrl) into a lentiviral expression vector, separately, and packaged them into lentiviral particles. The packaged lentiviruses were manufactured by cotransfection of HEK293T cells with Lipofiter (GeneChem Corp.). To construct stable AICDA-knockdown cell lines, 786-0 and 769-P cells in the logarithmic growth phase were seeded into 24-well plates containing 1 mL of complete media, at a concentration of 5000 cells per well. After 24 h of incubation, the shAICDA or shCtrl lentiviruses were added to the cells in the 24-well plates at a MOI = 100:1, separately. We then added 1 μL of polybrene per well. Twelve hours after infection, the medium was replaced with fresh complete medium. After 72 h, the cells were treated with 5 μg/mL of puromycin, and resistant colonies of 769-P or 786-0 cells were collected and expanded for further study.
Western blotting assay
Trypsin digestion of cells was followed by three washes with phosphate-buffered saline (PBS). The cells were then resuspended in Radioimmunoprecipitation assay buffer (Beyotime Biotechnology, Shanghai, China), and incubated with the protease inhibitor phenylmethanesulfonyl fluoride (Beyotime Biotechnology) and a protease inhibitor cocktail (Servicebio Corporation, Wuhan, China), on ice for ∼30 min. The cell lysates were further disrupted by ultrasonication before centrifugal separation at 12,000 g for 20 min at 4°C, after which the supernatant was collected. The protein concentration was measured using a bicinchoninic acid protein assay kit (Beyotime Biotechnology). The supernatant was then mixed with 0.25 × the volume of 5 × sodium dodecyl sulfate–polyacrylamide gel electrophoresis (SDS-PAGE) sample loading buffer (Beyotime Biotechnology), boiled at 100°C for 10 min to denature proteins, and samples of 20 μg of protein were separated on a 15% SDS-PAGE gel (Biotechwell, Shanghai, China), together with molecular weight markers (cat. no. 26617; Page Ruler Prestained Protein Ladder, Thermo, Lithuania). The separated proteins were then transferred to a polyvinylidene difluoride membrane (cat. no. IPFL00010; Millipore, Billerica, MA). The membranes were incubated for 1 h in 5% skim milk (cat. no. 232100; BD Biosciences, San Jose, CA) at room temperature before being washed three times with Tris-buffered saline (TBS). The membranes were then incubated with primary antibodies overnight at 4°C. After further washes, the membranes were incubated with secondary antibodies for 1 h at room temperature in the dark and then washed thrice with TBST (TBS with Triton-X-100). The immunoreactive proteins on the membranes were scanned using an Odyssey infrared imaging system (LI-COR Biosciences, Lincoln, NE). The experiments were repeated three times and analyzed using ImageJ software.
Quantitative real-time reverse transcription PCR
Total RNA from shCtrl and shAICDA ccRCC cells was extracted using the Trizol reagent (Invitrogen, Waltham, MA) according to the manufacturer's instructions. The concentration of the RNA was determined using ultraviolet spectrophotometry (Beckman Coulter, Indianapolis, IN). The cDNA was synthesized using a PrimeScript RT Reagent Kit with gDNA Eraser (RR047A; Takara, Shiga, Japan). Quantitative real-time PCR analysis for EMT marker mRNA levels was performed using a SYBR Premix Ex Taq II (RR820A; Takara) in an Applied Biosystems 7500 Real-Time PCR System (Thermo Fisher Scientific, Waltham, MA). Relative mRNA levels were calculated using the relative Ct method, and the fold change compared with the control (ACTB, encoding β-Actin). The primer sequences were as follows: E-cadherin forward: 5′-CTTTGACGCCGAGAGCTAC-3′, reverse: 5′-TTTGAATCGGGTGTCGAGGG-3′; N-cadherin forward: 5′-CCCCTCAAGTGTTACCTCA-3′, reverse: 5′-AAATCACCATTAAGCCGAGT-3′; Vimentin forward: 5′-TGCCGTTGAAGCTGCTAACTA-3′, reverse: 5′-CCAGAGGGAGTGAATCCAGATTA-3′; β-catenin forward: 5′-CATCTACACAGTTTGATGCTGCT-3′, reverse: 5′ GCAGTTTTGTCAGTTCAGGGA-3′; and β-actin forward: 5′ GTCCACCGCAAATGCTTCTA-3′, reverse: 5′- TGCTGTCACCTTCACCGTTC-3′.
Immunohistochemistry
AID was detected using IHC in tissue samples using antibodies against AID (1:25; Invitrogen). In brief, tissue sections were first deparaffinized using xylene and alcohol, and then rehydrated in buffer solution. A peroxidase sealing reagent was used to block endogenous peroxidase activity. An orthophoto microscope was used to view and image the tissue sections. The tissue sections were appraised and graded independently by two pathologists in a double-blinded manner.
Immunofluorescence
The renal cancer tissue sections were fixed in 4% paraformaldehyde and washed three times in PBS. After incubating in blocking buffer (5% BSA), primary antibodies against E-cadherin (1:50; Cell Signaling Technology, Danvers, MA), N-cadherin (1:200; Cell Signaling Technology), and AID (1:25; Invitrogen) were added to tissue sections and incubated at 4°C overnight. After rinsing three times with PBS, the tissue sections were incubated with secondary antibodies for 1 h at room temperature in the dark. After washing three times with PBS, the nuclei in the tissue sections were stained with antifade mounting medium including DAPI. Images were acquired under a fluorescence microscope (BX63; Olympus, Tokyo, Japan).
Assays for cell proliferation and colony formation
A Cell Counting Kit-8 (CCK-8) was used to assess cell proliferation according to the manufacturer's instructions (cat. no. CK04; Dojindo, Kumamoto, Japan). Polyclonal 769-P and 786-0 cells harboring shCtrl and shAICDA (2000 cells per well) were incubated in 96-well plates with 200 μL of fresh complete medium to explore AID's effect on ccRCC cell proliferation. At 0, 12, 24, 48, and 72 h, 10 μL of the water-soluble tetrazolium salt reagent was added to each well, and incubated with the cells for 1 h. Thereafter, the cells' absorbance at 450 nm was measured using a multiwell plate detector (Perkin Elmer, Singapore).
For the colony formation assay, polyclonal 769-P and 786-0 cells harboring shCtrl and shAICDA were seeded into 6-well plates at 500 cells per well, separately, and cultivated. Two weeks later, the cells were fixed in 75% alcohol for 30 min and stained with 0.05% crystal violet for 30 min. The colonies imaged under a microscope and calculated using the ImageJ software. The experiments were performed in triplicate to calculate the mean.
Wound healing assays
In six well plates, cells were grown to 80–90% confluence before a straight line wound was made in the confluent cell layer using a 1 mL plastic pipette tip. The wound was photographed at 0 and 24 h to assess the wound healing process:
The experiments were performed in triplicate to calculate the mean.
Cell invasion and migration assays
Twenty-four well transwell plates (cat. no. 3422; Corning, NY) lacking Matrigel (cat. no. 356234; BD Biosciences) in the lower chambers were used for the migration assay; however, for the invasion assay, the transwell plates were covered with Matrigel matrix at a 1:8 dilution ratio. 2 × 105 cells in 200 μL of RPMI 1640 medium without FBS were placed in the upper chamber, with 800 μL of RPMI 1640 containing 20% FBS being placed in the lower chamber. After incubation for 48 h at 37°C with 5% CO2, the cells that had migrated or invaded through the bottom chambers were fixed with 4% paraformaldehyde for 20 min and then stained with 0.05% crystal violet for 30 min. The amount of migrated or invaded cells was calculated by observation under a microscope. Each experiment was carried out in triplicate.
Statistical analysis
At least three biological repetitions were carried out for each experiment. The data are shown as the mean ± standard deviation. For experiments on AID-induced cell phenotypes, Student's t-test or one-way analysis of variance was used. All analyses were carried out using SPSS 23.0 (IBM Corp., Armonk, NY). A p-value <0.05 was considered statistically significant.
Results
Human renal cancer tissues express high levels of AID
Previously, AID was observed as highly expressed in many malignant tumors (Wright et al., 2015; Mohri et al., 2017; Araki et al., 2019). In this study, the level of AID was measured in ccRCC tissues and paracancerous tissues using Western blotting. The result indicated significantly higher levels of AID in tumor samples compared with that in paracancerous tissues (p < 0.001; Fig. 1A, B). We assessed AID using IHC in 36 clinical cases of ccRCC. IHC confirmed the increased level of AID in ccRCC tissues, providing a similar result to that of Western blotting (Fig. 1C, D).

Expression of AID, E-cadherin, and N-cadherin in ccRCC and paracarcinoma tissues.
HE staining was performed to compare the cell morphology and structure of tumor tissues and paracancerous tissues in six patients with ccRCC. Microscopically, the tumor tissue showed typical pathological findings, in which the cells had voluminous clear or eosinophilic cytoplasm, acinar/lamellar/tubular growth patterns, papillary or alveolar nested architecture, and an abundant vascular network. The tumor cell nuclei showed different degrees of atypia, such as prominent nucleoli, and the cells were spindle shaped. Individual cases showed transparent nodules and psammoma bodies with concentric patterns. The results showed that the normal renal tubular epithelium and renal parenchyma were severely damaged by the tumor cells showing high levels of AID (Fig. 1E).
In addition, in the primary lesions and adjacent normal kidney regions of six patients with ccRCC, the levels of AID, N-cadherin, and E-cadherin were assessed using immunofluorescence (Fig. 1F), which showed that ccRCC tissues had decreased levels of E-cadherin and increased levels of AID and N-cadherin, compared with those in para-ccRCC tissues. Interestingly, EMT was observed in the renal tumor tissues of almost all the patients, which suggested that the levels of AID, E-cadherin, and N-cadherin correlate closely in ccRCC tissues. Therefore, AID might be involved in the carcinogenesis and EMT of ccRCC.
AID knockdown in ccRCC cells
To identify ccRCC cell lines expressing high levels of AID, we assessed the AID level in ccRCC cell lines 769-P, 786-0, A498, and ACHN. The 769-P and 786-0 cells had the highest levels of AID (Fig. 2A, B), and were selected for subsequent study. Western blotting showed that the AID levels in 769-P and 786-0 cells were significantly higher than those in human renal proximal tubule epithelial cells (HK2) (p < 0.01, Fig. 2C, D), which provided further evidence that AID is involved in the formation of kidney cancer.

AID expression in ccRCC cells and AID knockdown in 769-P and 786-0 cells.
Next, AID expression was repressed in 769-P and 786-0 cells using two lentivirus-based RNA interference expression vectors (shAICDA1 and shAICDA2) in comparison with the nonsense shRNA as a negative control (shCtrl). Compared with those in shCtrl-769-P and shCtrl-786-0 cells, AID protein levels decreased by 32% and 29% in shAICDA2-769-P and shAICDA2-786-0 cells, respectively (p < 0.05; Fig. 2E–H), whereas AID levels decreased by 81% and 84% in shAICDA1-769-P and shAICDA1-786-0 cells, respectively. Obviously, shAICDA1 achieved the most significant repression of AID expression in 769-P and 786-0 cells (p < 0.001; Fig. 2E–H). Consequently, ccRCC cell 769-P and 786-0 expressing shAICDA1 were used in subsequent experiments.
AID, together with cell phenotype-regulating proteins, modulates EMT in ccRCC cells
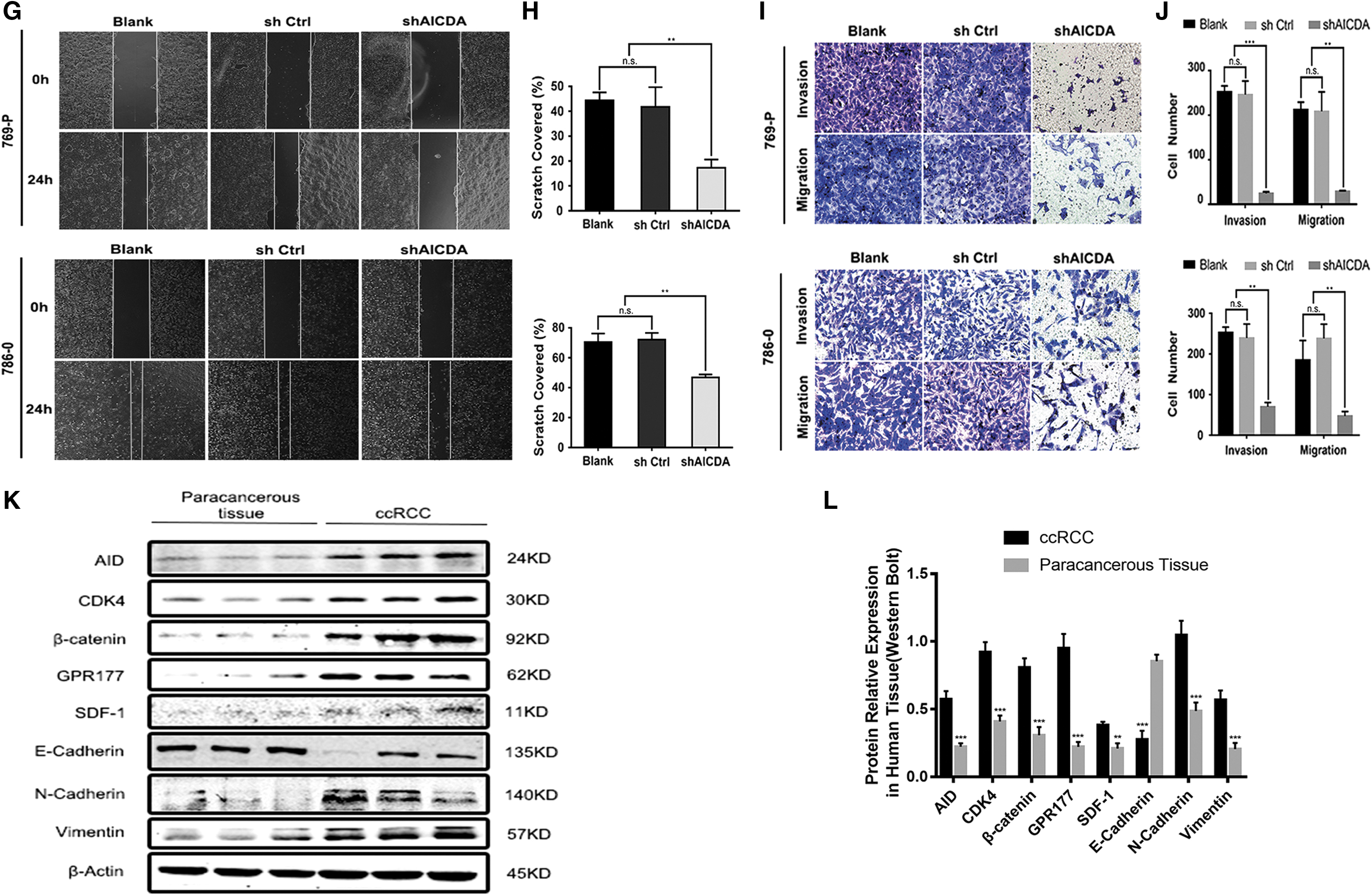
The expression levels of E-cadherin, N-cadherin, β-catenin, and Vimentin represent biomarkers for EMT (Song et al., 2017; Singh et al., 2018). Therefore, we measured the protein levels of EMT biomarkers in ccRCC samples. High levels of N-cadherin, β-catenin, and Vimentin, but low levels of E-cadherin, were detected in ccRCC tissues compared with those in para-ccRCC tissues (p < 0.001; Fig. 3K, L). Notably, the levels of N-cadherin, β-catenin, and Vimentin in the shAICDA group decreased visibly compared with those in the blank and the shCtrl groups. By contrast, the E-cadherin level increased significantly (p < 0.05; Fig. 3A–C). EMT can cause changes in invasion, migration, and proliferation of tumors, and AID targets include CDK4 cell-cycle-related protein, SDF-1 known as metastatic progression protein, and WLS associated with proliferation and metastasis (Yang et al., 2012; Hamaidi et al., 2019). Therefore, we assessed CDK4, WLS/GPR177, and CXCL12/SDF-1 levels to determine whether AID knockdown promotes EMT in ccRCC cell lines 769-P and 786-0. The protein levels of CDK4, CXCL12/SDF-1, and WLS/GPR177 decreased significantly in the shAICDA group compared with those in the blank and the shCtrl groups of 769-P and 786-0 cells (p < 0.05; Fig. 3A). Meanwhile, low levels of CDK4, CXCL12/SDF-1, and WLS/GPR177 were detected in para-ccRCC tissues compared with those in ccRCC tissues (p < 0.01; Fig. 3K, L). Taken together, these results demonstrated that repression of AID regulates many proteins involved in EMT, proliferation, and metastasis in ccRCC cells.
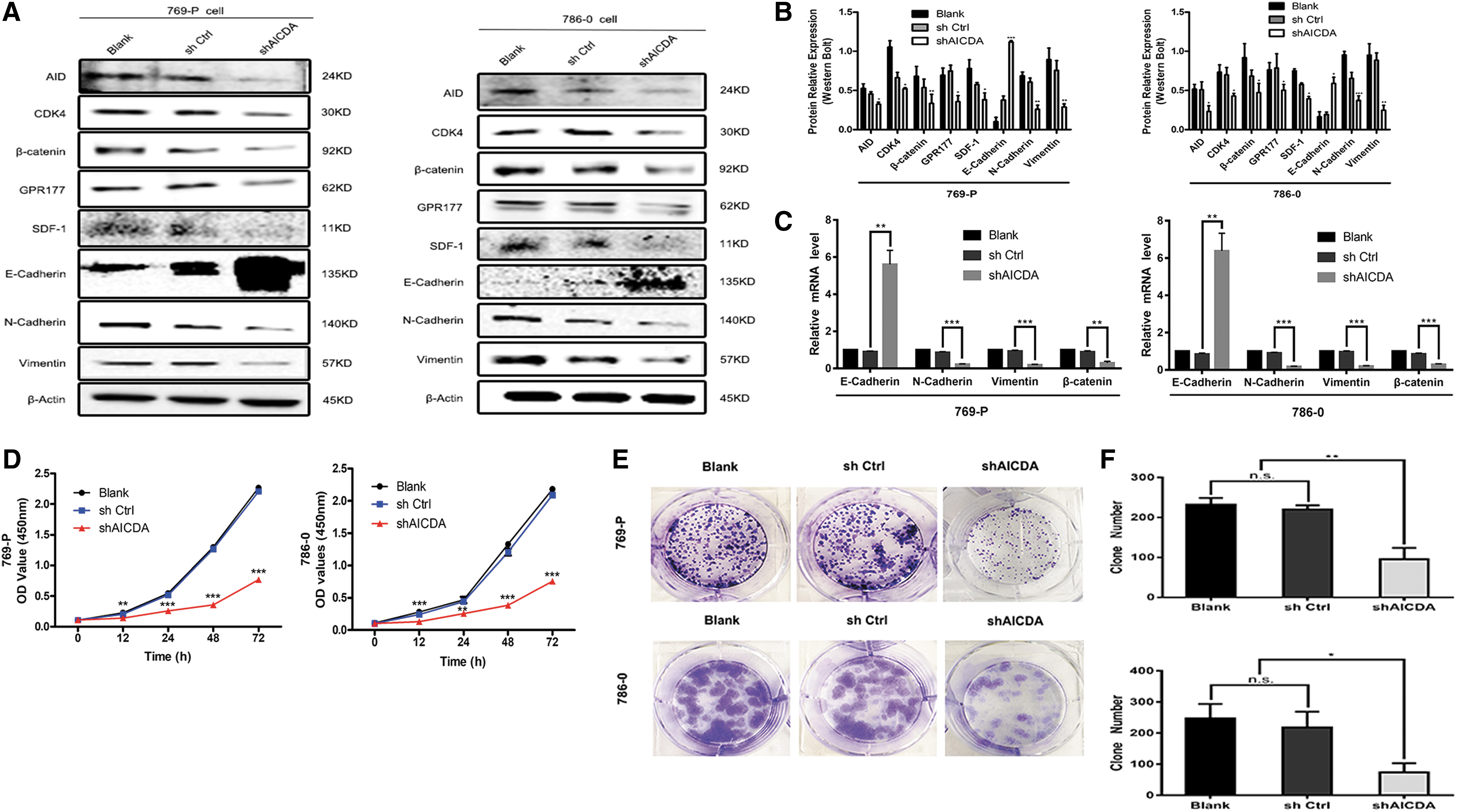
Effects of AID knockdown on EMT, downstream factors, and cell phenotype in ccRCC 769-P and 786-0 cells.
AID repression inhibits ccRCC cell migration, proliferation, and invasion in vitro
According to the above data, AID has a major role in EMT. In addition, ccRCC cells will undergo a series of biological functional changes after undergoing EMT. Therefore, CCK-8 assays were performed to assess the effect of AID repression during 769-P and 786-0 cell proliferation. CCK-8 assays were performed at 0, 12, 24, 48, and 72 h. The results showed that proliferation was significantly inhibited in the shAICDA group compared with that in the shCtrl and the blank groups of 769-P and 786-0 cells (p < 0.01; Fig. 3D). Similarly, the colony formation ability was remarkably decreased in the shAICDA groups, compared with that in the blank and the shCtrl groups of 769-P and 786-0 cells (p < 0.05; Fig. 3E, F). We then performed transwell and the wound healing assays to evaluate invasion and migration in response to AID knockdown in 769-P and 786-0 cells. As shown in Figure 3I–J, in the 769-P and 786-0 shAICDA groups, migration and invasion were significantly inhibited compared with that in the blank and shCtrl groups (p < 0.05). Similarly, slower migration in the wound healing assay was observed for the shAICDA group cells compared with that for the blank and shCtrl group cells (Fig. 3G, H). Thus, AID might promote the proliferation, migration, and invasion of ccRCC cells in vitro.
5-Azacytidine (DNA demethylation reagent) recovers the expression levels of WLS/GPR177, CXCL12/SDF-1, CDK4, and EMT, and the invasiveness in shAICDA ccRCC cells
AID modulates gene expression through active DNA demethylation (Bhutani et al., 2011). If DNA demethylation is deficient after AID knockdown, then the suppression of WLS/GPR177, CXCL12/SDF-1, and CDK4 should be recovered by treatment with the DNA demethylation reagent, 5-azacytidine (5-AZA or AZA). The levels of AID, WLS/GPR177, CXCL12/SDF-1, and CDK4, and those of EMT-related proteins E-cadherin, N-cadherin, Vimentin, and β-catenin were measured after AZA treatment. The results showed that AZA treatment significantly increased the levels of WLS/GPR177, CXCL12/SDF-1, and CDK4, and the EMT-related proteins, in the shAICDA group (p < 0.05); however, there was no change in the AID level (Fig. 4A, B). Furthermore, because of the toxicity of AZA, which inhibited cell growth to some extent, the effect on the proliferation of ccRCC cells was not obvious. Therefore, transwell assays were performed to observe AZA's effect on ccRCC cell invasion. Treatment with AZA recovered the invasion ability of 769-P and 786-0 cells with repressed AID expression (Fig. 4C, D). Taken together, the results suggested that AID might promote the expression of WLS/GPR177, CXCL12/SDF-1, and CDK4 through demethylation, and the observed changes in E-cadherin, N-cadherin, Vimentin, and β-catenin levels suggested that AID participates in EMT, whether it originates from AID directly or from the AID-WLS/GPR177 or AID-CXCL12/SDF-1 axes.

5-AZA promotes the expression of CDK4, GPR177, and SDF1 and increases the invasion of 769-P and 786-0 cells.

Discussion
AID is widely present in mammalian germinal center B lymphocytes. In recent years, AID has been widely studied because, as a DNA/RNA editing enzyme, it can induce point mutations and is closely implicated in human tumorigenesis, in which gene mutation is an important mechanism (Matsumoto et al., 2010). The aberrant expression of AID can induce cytosine deamination in genes, leading to C/G to T/A transitions, without C/G transversions or mutations at A and T (Martin et al., 2002; Shen et al., 2008). These mutations can lead to genomic DNA mutation and DNA double-strand breaks that promote the malignant transformation of cells. In addition, DNA demethylation could promote tumor initiation or progression through genomic instability and changes to gene expression. Demethylation by negative regulation caused the deletion of the tumor suppressor gene APC, which drives tumorigenesis (Andersen and Jones, 2013). Oncogenes, such as ELK1, FRAT2, R-RAS, RHOB, and RHO6, were identified as candidate genes that are repressed by DNA methylation in the normal stomach mucosa, whereas in a subset of gastric cancers DNA demethylation activates then (Nishigaki et al., 2005). AID-induced DNA demethylation might activate proto-oncogenes or eliminate tumor suppressor genes that promote tumor initiation or progression. However, the mechanism by which AID promotes tumor progression and the effect of AID on the tumor invasion, proliferation, and metastasis in ccRCC is unknown.
The results of this study demonstrated frequent AID overexpression in ccRCC tissues; however, it was barely detected in para-ccRCC tissue. Meanwhile, AID induced the expression of EMT-related markers. These results were consistent with expression levels of AID in 769-P, 786-0, and HK2 cells, and the results of previous studies for other malignant diseases, suggesting that high AID levels might be a potential diagnostic marker for ccRCC. To investigate the role of AID in ccRCC proliferation and metastasis, we repressed its expression in 769-P and 786-0 cells using a lentivirus-expressed shRNA sequence. AID repression resulted in decreased proliferation, invasion, and migration of ccRCC cells. EMT is a critical process in tumor cells invasion and metastasis (Munoz et al., 2013; Liu et al., 2019), which increases their motility and invasiveness (Hu et al., 2014). To confirm whether AID promotes ccRCC cell invasion and metastasis through the induction of EMT, we assessed the levels of EMT markers, E-cadherin, N-cadherin, Vimentin, and β-catenin in 769-P and 786-0 cells. In AID knockdown cells, E-cadherin levels were upregulated, and N-cadherin, Vimentin, and β-catenin levels were downregulated. The AID level correlated positively with N-cadherin, Vimentin, and β-catenin levels in ccRCC tissues, but negatively with E-cadherin levels. Thus, it seemed reasonable that the effects of AID on ccRCC cell invasion and metastasis might reflect its regulation of EMT.
Our previous study showed that AID-related progression factors, including WLS/GPR177, CXCL12/SDF1, and CDK4, are associated with bladder uroepithelium cell carcinoma (BUCC) (Li et al., 2019); however, the relationship between AID and these factors in ccRCC was unknown. WLS/GPR177, localizing to the Golgi, endocytic compartments, and the plasma membrane (MacDonald et al., 2009), is an essential seven-pass transmembrane protein that binds the cytoplasmic WNT ligand and secretes it to the cytomembrane, mediating the classical Wnt/β-catenin pathway (Fu et al., 2009; Seo et al., 2018). High expression of WLS promotes proliferation and metastasis of ccRCC cells (Yang et al., 2012; Xu et al., 2016; Schmid et al., 2017). β-Catenin, a major component of adherens junctions, links the actin cytoskeleton with the transmembrane molecule E-cadherin, playing an important role in EMT (Castaneda et al., 2017). Meanwhile, as a key molecule in the Wnt/β-catenin pathway, β-catenin is dephosphorylated and translocates to the nucleus, where together with T-cell factor/lymphoid enhancer transcription factors, activates Wnt target gene expression (including c-Myc, cyclin D1, and matrix metalloproteinase 7) (Yang et al., 2018), which is implicated in EMT and carcinogenesis (Zhang et al., 2014; Schmid et al., 2017). Hence, AID might modulate the Wnt/β-catenin signaling pathway through WLS. CDK4 functions in mammalian cell proliferation, driving cells to progress into the S phase of the cell cycle. CDK4's enzyme activity in the G1 phase is regulated by D-type cyclins (e.g., cyclin D1), which are expressed in response to various extracellular signals, such as inhibitory cytokines, stimulatory mitogens, cell–cell contacts, differentiation inducers, and other spatial cues (Sherr et al., 2016). Increased expression of CDK4 might be regulated by the Wnt/β-catenin pathway to strengthen the cell proliferation of ccRCC cells (Hseu et al., 2016). The chemokine SDF1/CXCL12 is expressed by stromal cells (Bouyssou et al., 2016), has crucial functions in angiogenesis and embryogenesis, and is associated with tumor metastasis and the mediation of EMT (Hu et al., 2014). Increased expression of SDF1 might also be regulated by Wnt/β-catenin pathway to promote the metastasis of ccRCC cells (Hamaidi et al., 2019). The Wnt/β-catenin signaling pathway has an important function in the induction of EMT and the malignant progression of carcinomas (Qi et al., 2016; Xie et al., 2019). The results of this study demonstrated that AID knockdown caused the downregulation of WLS, β-catenin, CDK4, and SDF-1 and suppressed the invasion and metastasis of ccRCC cells. Thus, AID might directly regulate EMT, CDK4, and SDF1, possibly through the WLS/Wnt/β-catenin signaling pathway. However, 5-AZA, a DNA demethylation reagent, recovered the expression levels of WLS and CXCL12 in ccRCC cells, but did not recover their expression in BUCC (data not shown) (Supplementary Data) (Li et al., 2019). The level of regulation complexity and the importance of the Wnt/β-catenin signaling pathway strongly suggest that these issues should be further explored in future studies.
In summary, this study showed that elevated levels of AID were closely related to tumorigenesis in patients with ccRCC, and that AID repression could suppress cell proliferation, migration, and invasion of ccRCC cells. Furthermore, AID might promote WLS, CDK4, and SDF1 expression through demethylation. Previous studies showed that WLS and SDF1 facilitate EMT and metastasis in many malignant diseases, which is probably regulated by AID directly or through the AID–WLS axis. Nevertheless, WLS and SDF-1 upregulation might promote the invasiveness of 769-P and 786-0 cells, and be in turn regulated by AID through demethylation. The toxicity of 5-AZA meant that we could not clearly determine the effects of CDK4 and WLS on the proliferation of ccRCC cells. Therefore, AID might promote ccRCC progression through WLS/GPR177, CXCL12/SDF1, and CDK4, particularly during metastasis and proliferation; however, the specific regulatory mechanism requires further study. AID also represents a potential clinical therapeutic target to treat metastatic ccRCC.
Footnotes
Acknowledgments
We thank Yuqi Xia, Qi Wu, Siqi Zhou, and Wenyi Jin for their brilliant technical assistance and Zhihua Wang for his contribution to this article.
Disclosure Statement
The authors declare that they have no conflict of interest.
Funding Information
This work was supported by National Natural Science Foundation of China (Grant No. 81360375), Natural Science Foundation of Hainan province (Grant No. 818QN317), and special fund project of Science and Technology of Social Development of Hainan province (Grant No. ZDYF2019122).
Supplementary Material
Supplementary Data
References
Supplementary Material
Please find the following supplemental material available below.
For Open Access articles published under a Creative Commons License, all supplemental material carries the same license as the article it is associated with.
For non-Open Access articles published, all supplemental material carries a non-exclusive license, and permission requests for re-use of supplemental material or any part of supplemental material shall be sent directly to the copyright owner as specified in the copyright notice associated with the article.