
Abstract
Wnt signaling is activated in many cancer types, yet targeting the canonical Wnt pathway has been challenging for cancer therapy. The pathway might be effectively targeted at many levels depending on the mechanism by which it has become hyperactive. Recently, mouse genetic screens have found that R-spondins (RSPOs) act as oncogenes. Evidence includes recurrent genomic rearrangements that led to increased RSPO2 or RSPO3 expression in human colorectal adenocarcinomas, exclusive of APC mutations. RSPOs modulate Wnt signaling to promote epithelial cell proliferation and survival. These secreted proteins modulate Wnt signaling by binding to G-coupled receptors LGR4/5/6, ultimately inhibiting frizzled membrane clearance by RNF43 and ZNRF3. They also exert their function independent of leucine-rich repeat-containing, G protein-coupled receptors (LGRs) by binding to ZNRF3 and RNF43. This results in increased β-catenin concentration that, after translocation to the nucleus, acts as a transcriptional coactivator of genes necessary for proliferation and cell survival. In this article, we aimed to identify the role of RSPOs in colon and breast cancers by using in silico and in vitro studies. We found that expression of RSPO2 and RSPO3 at high levels characterized a subset of colorectal cancers (CRCs). RSPO2 expression was found to characterize a subset of triple-negative breast cancers. In both instances, increased expression of RSPOs was associated with an activated Wnt signaling gene expression profile. Furthermore, knockdown of RSPO2 decreased Wnt signaling and proliferation in human breast cancer cells. Our findings show and confirm that RSPO2 and RSPO3 expression is upregulated in a subset of colorectal adenocarcinomas and breast cancers and that both are attractive druggable oncoprotein targets against such cancers. We also describe novel fusion transcripts that occur in CRC.
Introduction
Activated canonical Wnt signaling is a feature of many types of cancer and has, subsequently, generated significant interest in targeting the Wnt pathway therapeutically. Multiple strategies for targeting the Wnt pathway have been developed, such as porcupine and tankyrase inhibitors with limited success in clinical trials (Anastas and Moon, 2013). Because Wnt signaling is integral for regulation of normal cell development and homeostasis, it is difficult to avoid off-target effects of systemic therapies aimed at reducing Wnt signaling (Barker and Clevers, 2006; Kahn et al., 2014). Therefore, an ideal target would be a cancer-specific activator of Wnt signaling.
In cancer, multiple genetic events are known to activate Wnt signaling. For example, in colorectal cancer (CRC), the predominant mechanism of Wnt pathway activation is an inactivating mutation or deletion of APC, a core negative regulator of CTNNB1. Activating mutations in CTNNB1, loss of function mutations in AXIN1/2, and other pathway components are alternative routes to Wnt activation in CRC (Fodde et al., 2001). In breast cancer, activation of Wnt signaling has been correlated with the basal subtype, but the events leading to Wnt activation in breast cancer are incompletely understood (Khramtsov et al., 2010). Silencing of SFRP1, a secreted negative regulator of the Wnt pathway, is one proposed mechanism that promotes Wnt signaling in breast cancer (Ugolini et al., 2001).
R-spondins (RSPOs) have been described as secreted factors that modulate Wnt signaling. They are thought to modulate Wnt signaling by binding to LGR4/5/6 (de Lau et al., 2012). They have also been found to exert their function independently of LGRs, by binding directly to RNF43 and ZNRF3 (Szenker-Ravi et al., 2018). RSPOs were proposed as human oncogenes in 4–10% CRC cases that showed RSPO2 and RSPO3 overexpression, due to recurrent genomic rearrangements, in the absence of APC inactivating mutations (Seshagiri et al., 2012; Shinmura et al., 2014). Studies in mice also support the hypothesis that RSPOs are oncogenic and modulate Wnt signaling. Insertional mutagenesis screens performed in Apc wild-type mice identified Rspo2 as a common insertion sites in gastrointestinal tract colorectal-like tumors (Lowther et al., 2005; Theodorou et al., 2007; Starr et al., 2009; Takeda et al., 2015). Moreover, murine mammary tumor virus screens identified Rspo2 and Rspo3 as breast tissue oncogenes (Lowther et al., 2005; Theodorou et al., 2007).
Interestingly, similar screens using Apc mutant mice did not identify RSPOs and the same exclusivity between RSPO2/3 activation and APC mutation was also found in human CRC, suggesting that activation of RSPOs can substitute for loss of APC to drive Wnt signaling (March et al., 2011; Starr et al., 2011; Seshagiri et al., 2012). Functional studies in mice demonstrated that targeted overexpression of Rspo1 in the intestines promotes hyperplasia, whereas overexpression of Rspo2 in mammary epithelium promotes breast cancer (Kim et al., 2005; Klauzinska et al., 2012). Other studies have also shown that RSPO2 overexpression in its native form and in a fusion transcript activates Wnt signaling and induces colorectal tumors (Han et al., 2017).
One mechanism proposed for sustained Wnt signaling through RSPO/LGR interaction is through inhibition of a negative feedback loop. Activation of Wnt signaling results in transcription of ZNRF3, an E3 ubiquitin ligase, that translocates to the cell membrane, ubiquitinates the frizzled (FZD) receptor, and dampens Wnt signaling in a canonical negative feedback loop. Recent studies demonstrated that RSPO1 and LGR4 together can cause membrane clearance of ZNRF3, breaking the negative feedback loop, resulting in enhanced Wnt signaling (Hao et al., 2012; Koo et al., 2012). Surprisingly, Wu et al. (2014) demonstrated the opposite effect when analyzing RSPO2 and LGR5 in certain CRC cell lines. They found that RSPO2 and LGR5 stabilize ZNRF3 at the surface, resulting in diminished Wnt signaling. Supporting their hypothesis that RSPO2 acts as a tumor suppressor, they found that the majority of CRC patients have downregulated RSPO2 through promoter methylation. These findings indicate the RSPOs have context-dependent pleiotropic effects. A clear analysis of expression data on RSPO2 and RSPO3 levels and canonical Wnt signaling-induced gene expression has not been reported for human cancer.
In this study, we focused on the role of RSPO2 and RSPO3 in two major epithelial cancers: colon and breast. We present evidence that RSPO2 and RSPO3 may function as oncogenes in a relatively small subset of these cancers based on analysis of expression levels, presence of RSPO fusion genes, and in vitro functional studies.
Methods
Acquisition of RNA-seq and somatic mutation data from The Cancer Genome Atlas
RNA-seq and somatic mutation data were extracted from The Broad Institute GDAC Firehose. These data were generated by The Cancer Genome Atlas (TCGA) Research Network. RNA-seq normalized counts (RSEM) were obtained for a set of 69 genes of interest related to RSPOs, Wnt signaling, and tissue-specific markers of differentiation and stemness, for 1884 samples comprising 41 normal colon, 434 CRCs, 111 normal breasts, and 1048 breast carcinomas. Somatic mutation data were obtained for APC, CTNNB1, RSPO1, RSPO2, RSPO3, and RSPO4 from 266 CRCs and 768 breast carcinomas.
Primary breast tissue RNA isolation, cDNA synthesis, and quantitative reverse transcriptase polymerase chain reaction
Normal/tumor matched pairs were obtained from the University of Minnesota Tissue Procurement facility. In brief, cDNA synthesis and qPCR were performed as described (Burns et al., 2013). Tissue RNA was from 100 mg flash-frozen tissue disrupted by a 2-h water bath sonication in 1 mL of Qiazol Lysis Reagent (RNeasy, Qiagen). Cell RNA was made using Qiashredder (RNeasy, Qiagen). qPCR was performed on a Roche Lightcycler 480 instrument. RSPO2 and RSPO3 primer sets were designed using the ProbeFinder version 2.48 for the Human Universal ProbeLibrary (UPL) from Roche Applied Science. The housekeeping gene TBP was used for normalization. Primer and probe sequences are listed in Supplementary Table S2.
Tissue culture reagents and cell lines
BT549, MCF10A, and MCF7 cells were obtained from the American Type Culture Collection (ATCC). BT549 and MCF7 cells were cultured in Roswell Park Memorial Institute (RPMI) 1640 medium supplemented with 10% fetal bovine serum and 0.023 IU/mL bovine insulin. MCF10A cells were cultured in Dulbecco's modified Eagle medium (DMEM) supplemented with 5% horse serum, 20 ng/mL epidermal growth factor (EGF), 0.5 μg/mL hydrocortisone, 100 ng/mL cholera toxin, 10 μg/mL insulin, and 1% penicillin/streptomycin (Debnath et al., 2003). All cells were grown on tissue culture-treated plates under standard conditions of 37°C and 5% CO2.
In vitro gene knockdown and overexpression and proliferation assays
For RSPO2 knockdown experiments, plasmids encoding lentiviral shRNAmirs against RSPO2 or a nonsilencing control shRNAmir were purchased from OpenBiosystems. Lentiviral particles were produced in 293T cells using the Trans-Lentiviral Packaging Kit (Thermo Scientific). For overexpression experiments, lentiviral expression vectors were cloned with RSPO2 or dsRed regulated by a CAGGs promoter and followed by an IRES-GFP to monitor transduction efficiency. Lentiviral particles were produced in 293T cells by cotransfection with helper plasmids. For both knockdown and overexpression experiments, viral supernatant was collected after 24 h of virus production, cleared, and applied to transduce experimental cells with 12 μg/mL polybrene overnight. Transduced cells were selected with 1 μg/mL puromycin. Knockdown efficiency and overexpression levels were assayed by quantitative reverse transcriptase polymerase chain reaction (qRT-PCR).
Quantitative reverse transcriptase polymerase chain reaction
RNA was isolated from cell lines using the Trizol method (Thermo Scientific). RNA samples were treated with DNase to remove contaminating genomic DNA (Turbo DNA-free Kit, Ambion). Complementary DNA was synthesized from 1 μg template RNA per sample using random hexamer primers (SuperScript III First-Strand Synthesis System, Invitrogen). qRT-PCRs were conducted with FastStart Universal SYBR Green Master mix (Roche), using 0.5 μL of cDNA template per 25 μL reaction. Primer sequences for qRT-PCRs are listed in Supplementary Table S2. Data were analyzed by normalization to ACTB using the following equation: relative expression = ((2^(CT_ACTB))/(2^(CT_GOI))).
Results
RSPO2 and RSPO3 are upregulated in fusion-transcript-positive colon cancer
To determine the frequency of RSPO overexpression and gene rearrangement, we analyzed RNA-seq data from 434 CRCs and 41 normal colon samples obtained through TCGA. We focused on RSPO2 and RSPO3 as previous data by Seshagiri and colleagues suggested that RSPO2 and RSPO3 are CRC drivers. Also, RSPO2 and RSPO3 have been shown to be the more potent Wnt modulators of the four RSPOs (Moad and Pioszak, 2013). In this cohort, RSPO2 and RSPO3 expression was decreased in the majority of CRCs compared with normal colon. Specifically, 422 of 434 (97.2%) CRCs have more than fourfold decreased RSPO2 expression, and 250 of 434 (57.6%) have more than fourfold decreased RSPO3 expression (Fig. 1A). This concurs with previous observations indicating RSPO2 and RSPO3 expression levels are suppressed in the majority of CRCs (Kazanskaya et al., 2004; Seshagiri et al., 2012; Shinmura et al., 2014; Wu et al., 2014).
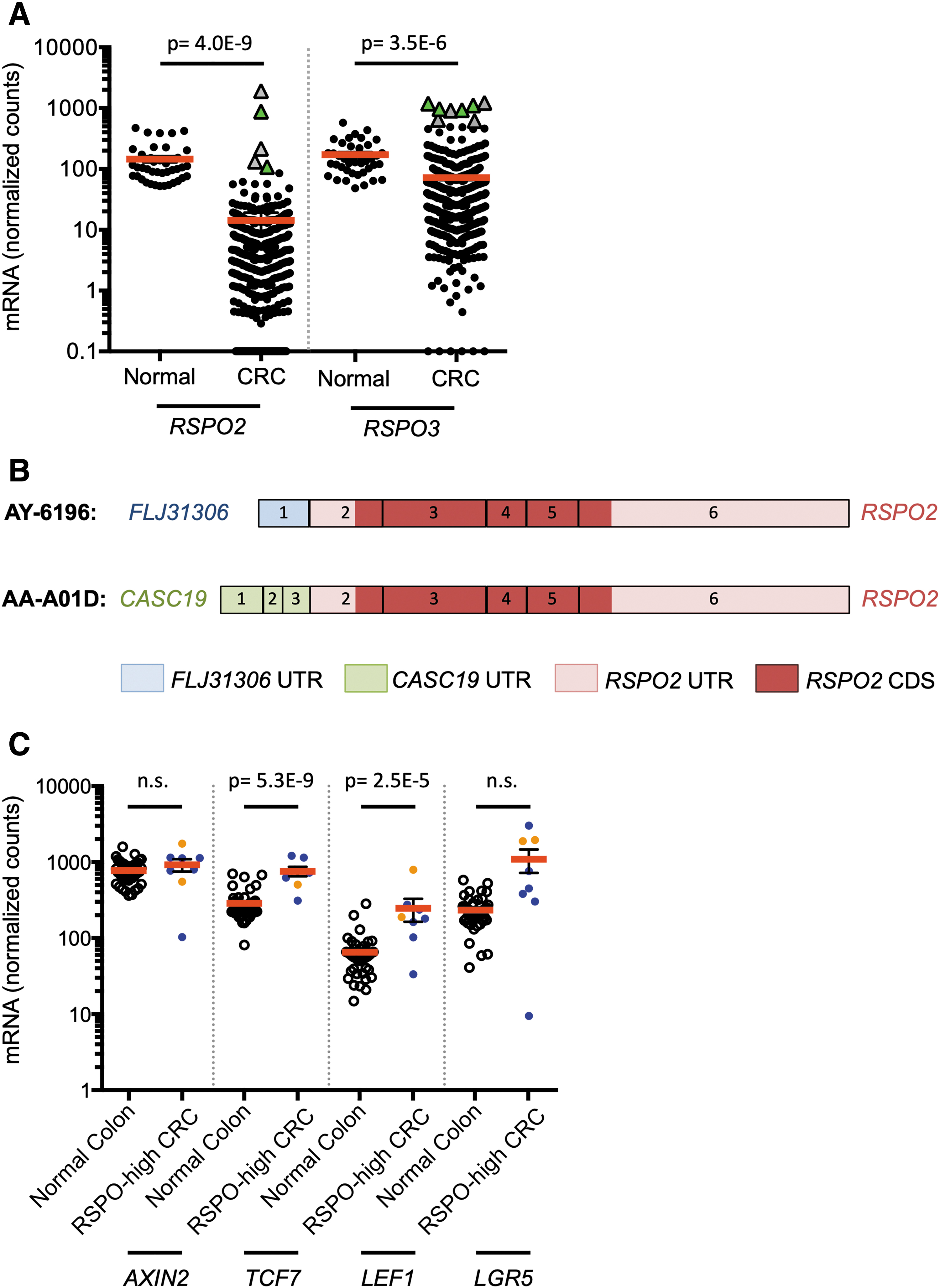
RSPO2 and RSPO3 are highly expressed in rare colorectal tumors and associated with expression of RSPO fusion transcripts and activated Wnt signaling.
Notably, a small subset of CRCs expressed high levels of RSPO2/3. “RSPO-high” tumors were defined as having greater than fourfold elevated mRNA levels compared with normal colon. In this cohort, two CRCs (0.5%) were RSPO2-high tumors and six CRCs (1.4%) were RSPO3-high tumors (Fig. 1A). Compared with the corresponding RSPO-low tumors, RSPO-high tumors expressed 231-fold higher RSPO2 levels and 59-fold higher RSPO3 levels.
Next, we sought to determine whether RSPO-high samples expressed RSPO gene fusions. Tumors that had paired-end RNA-seq data available (three of eight) were analyzed with DeFuse (McPherson et al., 2011). All three expressed PTPRK-RSPO3 fusion transcripts similar to prior studies (Storm et al., 2016) (Fig. 1A). An additional novel FLJ31306-RSPO2 fusion was found in a sample with elevated RSPO2 expression less than the cutoff for RSPO2-high designation. We used Trinity (Grabherr et al., 2011) to locate RSPO fusion transcripts in samples with single read RNA-seq data, and to confirm the FLJ31306-RSPO2 fusion. This method identified one additional PTPRK-RSPO3 fusion in an RSPO3-high sample, and a novel CASC19-RSPO2 fusion in an RSPO2-high sample (Fig. 1B and Supplementary Fig. S1). The CASC19-RSPO2 and FLJ31306-RSPO2 fusions contained the native start codon in RSPO2 exon 2 and were predicted to overexpress the complete native coding sequence, similar to the previously described EIF3E-RSPO2 fusion (Seshagiri et al., 2012). Examination of RSPO2 mRNA levels on a per exon basis revealed a pattern of exon imbalance, further suggesting that the high levels of RSPO2 mRNA in these samples consisted of fusion transcripts, as did the RSPO3 fusion (Supplementary Fig. S1 and Supplementary Table S1). These data show that RSPO2 can be activated by fusion with genes other than EIF3E.
RSPO2/3-high human CRCs have activated Wnt signaling and wild-type APC
To determine the relationship between RSPO overexpression and Wnt signaling, we next examined expression of a panel of Wnt-regulated genes (AXIN2, TCF7, LEF1) and LGR5, a Wnt target gene that is also the receptor for RSPOs. In the TCGA set of CRC samples, expression of three of four Wnt target genes was increased in RSPO-high tumors compared with normal colon (Fig. 1C). Consistent with prior reports, RSPO-high status was mutually exclusive with APC mutations (Seshagiri et al., 2012; Shinmura et al., 2014). Specifically, among RSPO-high CRCs with available somatic mutation data, four of four (100%) retained wild-type APC. These data are consistent with a model wherein RSPOs are an alternative route to activation of Wnt signaling.
It has been proposed that in CRC with an RSPO gene fusion causing overexpression, LGR5 must be downregulated to prevent Wnt inhibition through a repressive RSPO/LGR5 axis (Wu et al., 2014). In contrast, we found that LGR5 mRNA was downregulated in only one of eight RSPO-high CRCs, and was upregulated in the other seven samples, consistent with other Wnt-responsive genes (Fig. 1C). These data suggest that LGR5 inactivation is not a requirement for Wnt activation in RSPO-high tumors. Because Wnt activation can promote tumorigenesis in other tissues, we next examined expression of RSPO2 and RSPO3 in primary human breast cancer, using TCGA data sets and independently analyzed primary samples.
RSPO2 is upregulated in human breast cancer and associated with basal/HER2 subtypes and active Wnt signaling
To analyze RSPO expression in breast cancer, we obtained pairs of matched normal and tumor tissues from 41 patients. RSPO2 and RSPO3 mRNA expression levels were quantified by qRT-PCR. Six tumors (14.6%) expressed RSPO2 mRNA levels more than fourfold higher than their matched normal controls and were defined as “RSPO2-high” (range: 6.7 to 28.2-fold elevation) (Fig. 2A). RSPO2-high breast tumors did not express EIF3E-RSPO2 fusion genes by RT-PCR. In the TCGA data set, we compared 1048 breast tumors with 111 normal breast tissues. RSPO2 mRNA was expressed at a very low level in normal breast samples (normalized counts <1 in 100 of 111 samples, 90.1%). The majority of breast tumors also maintained low RSPO2 expression (normalized counts <1 in 842 of 1048 samples, 80.3%). However, 122 of 1048 (10.6%) of breast tumors had >4-fold increased RSPO2 expression compared with the normal average (range: 4.0 to 300.7-fold elevated) (Fig. 2B). Similar to our observations in CRC, RSPO3 levels were decreased in the majority of breast tumors, both in the set of 41 matched tumor/normal samples and in the TCGA data set (Fig. 3A and B). In the TCGA data set, 881 of 1048 (83.5%) of breast tumors were RSPO3-low, whereas rare tumors had elevated RSPO3 levels (3 of 1048, 0.3%, Fig. 3B). DeFuse analysis of select RSPO-high breast tumors in the TCGA set did not identify expression of RSPO fusion transcripts (Table 1).
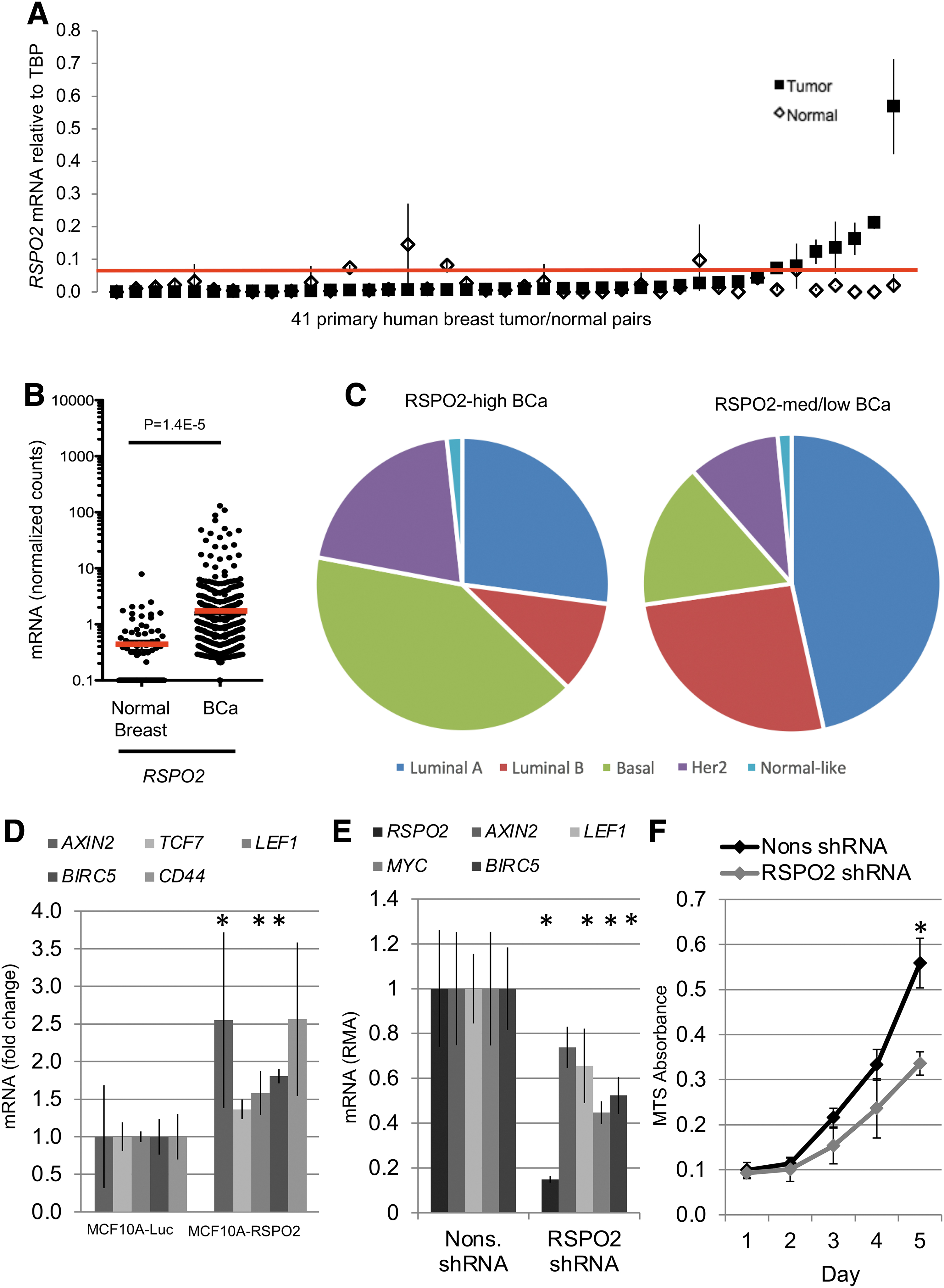
RSPO2 is highly expressed in a subset of breast tumors and associated with the basal and HER2 subtypes.
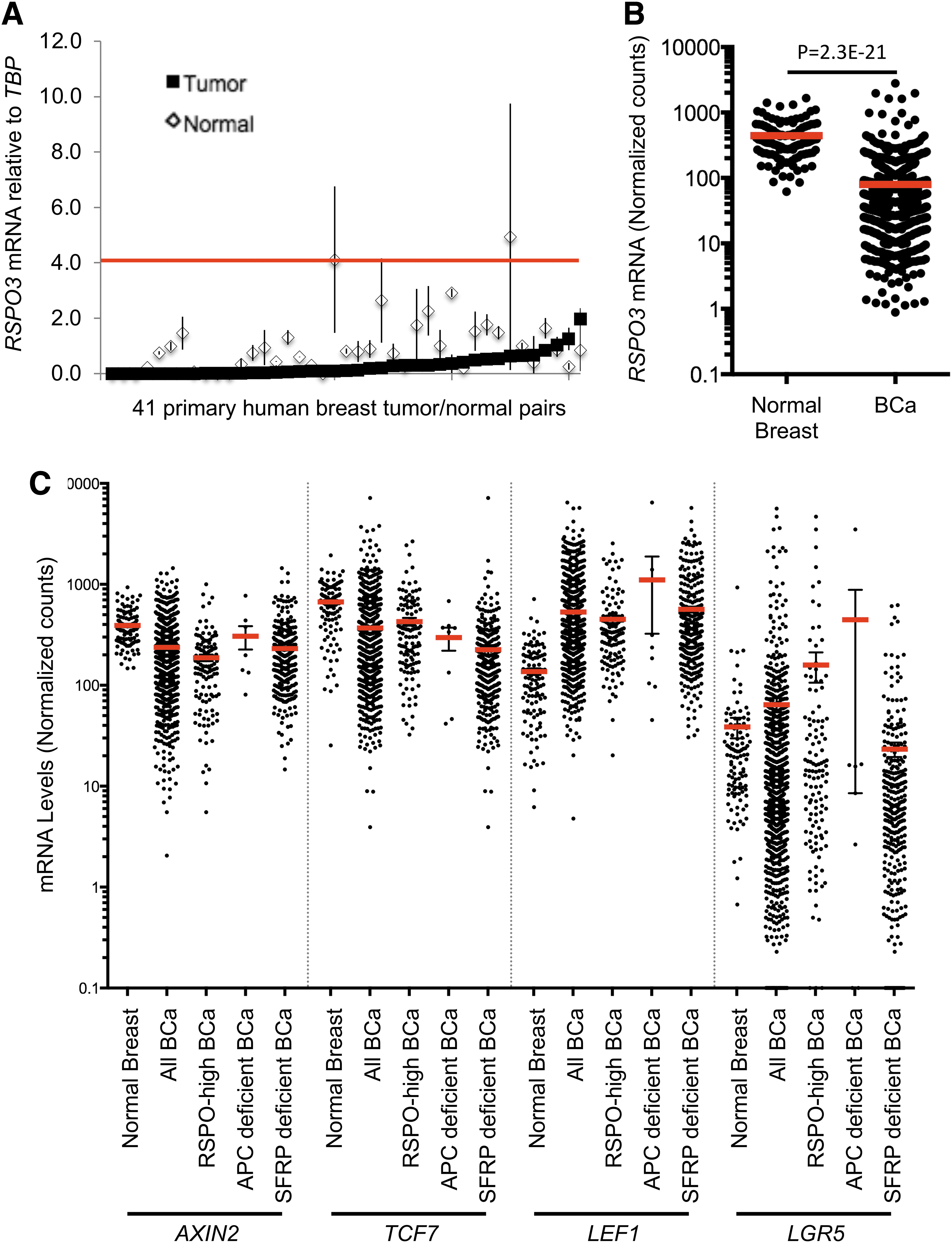
Breast cancers have decreased expression of RSPO3 and mixed expression of Wnt target genes.
Summary of Fusion Gene Analyses
N/A, not analyzed; nd, not detected; TCGA, The Cancer Genome Atlas.
To determine the clinical relevance of RSPO2 overexpression, we next analyzed PAM50 molecular subtype information for 521 breast tumors in the TCGA data set. We also looked into RSPO3 levels in matched human breast cancer samples (Fig. 3A) and RNAseq data in TCGA (Fig. 3B). Interestingly RSPO3 expression in human tumors is lower than in normal adjacent tumor. RSPO2-high status was significantly associated with basal (p = 2.07E-5) and HER2 (p = 0.0259) subtypes, and anticorrelated with luminal A (p = 0.0052) and luminal B (p = 0.0093) subtypes (Fig. 2C). Significantly, although basal-type tumors account for only 19% of tumors overall, 41% of RSPO2-high breast tumors were basal-type tumors (Fig. 2C).
Although active Wnt signaling, as measured by presence of nuclear CTNNB1, is associated with basal subtype tumors (Khramtsov et al., 2010; Geyer et al., 2011), expression of Wnt target genes in RSPO-high breast tumors was not consistently elevated compared with normal tissue (Fig. 3C and Supplementary Fig. S3). Specifically, although expression levels of the Wnt target genes LEF1 and LGR5 were significantly elevated in RSPO-high tumors, those of AXIN2 and TCF7 were significantly downregulated (Fig. 3C). Examination of Wnt target gene expression in breast tumors with APC loss of function mutations or reduced expression of SFRP1 revealed a similar pattern, suggesting that Wnt signaling is equivalently activated in these subsets (Fig. 3C).
RSPO2 regulates Wnt signaling and proliferation in breast cancer cells
To determine the functional significance of elevated RSPO2 expression in breast cancer, we overexpressed RSPO2 in a nontransformed basal-type breast epithelial cell line (MCF10A) (Coussy et al., 2017), and we knocked down RSPO2 in BT-549, a RSPO2-high basal-type breast cancer cell line. MCF10A cells overexpressing RSPO2 showed transcriptional upregulation of Wnt target genes (Fig. 2D). Wnt target genes were selected based on data gathered and published by the Nusse laboratory's Wnt Homepage. BT-549 cells express extremely high levels of RSPO2 compared with 58 breast cancer cell lines profiled in the Cancer Cell Line Encyclopedia (Fig. 3A). Knockdown of RSPO2 in BT-549 cells resulted in decreased expression of Wnt target genes, as well as reduced cell proliferation (Fig. 2E, 2F, and Supplementary Fig. S2). These results suggest that RSPO2-high breast cancers may require RSPO2 expression for activation of Wnt signaling and enhanced growth.
Discussion
The role of RSPOs in regulating the Wnt pathway in cancer is complex, as evidence suggests that RSPOs can function as oncogenes or tumor suppressors (Seshagiri et al., 2012; Wu et al., 2014). To clarify the role of RSPOs, specifically RSPO2 and RSPO3, we analyzed gene expression, the presence of fusion transcripts, and functional effects of RSPO2 and RSPO3 in colon and breast cancers. We found that, in a minority of cases of both cancers, RSPO2 or RSPO3 are significantly upregulated and may play a functional role enhancing Wnt signaling and promoting tumor development in these cancers.
Colon cancer
In a majority of colon cancers, RSPO2 expression is suppressed due to promoter methylation. Wu et al. (2014) demonstrated that RSPO2 along with LGR5 causes stabilization of ZNRF3, an E3 ubiquitin ligase that negatively regulates Wnt signaling. They proposed that loss of RSPO2 in CRC patients would lead to increased Wnt signaling through destabilization of ZNRF3. In a small subset of CRC patients, however, RSPO2 or RSPO3 is highly overexpressed due to gene fusions, such as PTPRK-RSPO3 and EIF3E-RSPO2, which result in changes in promoter regulation (Seshagiri et al., 2012; Shinmura et al., 2014). We extended this finding by analyzing TCGA RNA-Seq data for gene fusions. We found gene fusions in 66% of RSPO3-high cancers and 50% of RSPO2-high cancers. In addition to previously identified fusions, we detected two novel RSPO2 gene fusions that also contained the native start codon of RSPO2, similar to the EIF3E-RSPO2 fusion.
Moreover, we identified CASC19-RSPO2 and FLJ31306-RSPO2 fusion genes to be enriched in a subset of CRCs. These new data suggest that there are many circumvented pathways that result in the overexpression of RSPO2, resulting in positive Wnt signaling. Such events may be as a result of loss of border elements, such as CCCTC-binding factor (CTCF) resulting in aberrant chromatin confirmation, resulting in fusion transcript generation, enhancer hijacking, and/or transcription run-off. Analysis of exon-level expression confirmed that high-expression levels in these cancers were likely due to transcription of the fusion transcript. In the report by Wu et al., it was suggested that elevated levels of RSPO2 would only modulate Wnt signaling if LGR5 levels were downregulated. In the TCGA data set, however, we found that LGR5 was upregulated in 87% (7/8) of RSPO2/3 high cancers, indicating loss of LGR5 is not necessary for RSPO-induced Wnt activation. These findings support a model in which RSPO2 or RSPO3 is upregulated in a subset of APC wild-type CRCs due to gene fusions or other events, correlating with activation of Wnt signaling.
Breast cancer
Similar to colon cancer, a majority of breast cancers have constitutively low levels of RSPO2 and RSPO3 expression. However, we found that a small subset has elevated RSPO2 levels and this subset is enriched for the basal subtype of breast cancer. We manipulated levels of RSPO2 by overexpression in an RSPO2-low breast epithelial cell line (MCF10A), which resulted in elevated levels of Wnt target genes. Decreasing levels of RSPO2 using shRNA in an RSPO2-high cell line (BT-549) caused a decrease in expression of Wnt target genes and a decrease in proliferation. These functional studies indicate that RSPO2 can function as an enhancer of Wnt signaling in breast cancer. The mechanism of upregulation of RSPO2 in breast cancer is unclear, as we did not detect fusion transcripts, nor did we detect a positive feedback loop through Wnt3a treatment, as proposed by Kazanskaya et al., (2004). Moreover, others have found that increasing RSPO2 expression in breast tumors results in decreased metastasis-free survival (Coussy et al., 2017) (Supplementary Fig. S4). Further studies will be required to determine the mechanisms of RSPO upregulation and characterize Wnt signaling role and function in breast cancer to find sensible drug target.
We have found and confirmed that RSPOs are overexpressed in a small subset of human cancers of the colon and breast. This occurs exclusive of APC mutations and other Wnt pathway component alterations. Many genetic mechanisms are involved in the increased expression of RSPOs. Genomic rearrangements that result in fusion transcript expression, copy number variations, and epigenetic alterations that control proper promoter function have been identified. It is important then to further study RSPOs as druggable targets and biomarkers of human cancers, with a new focus on breast cancer (Chartier et al., 2016; Storm et al., 2016; Coussy et al., 2017). Alternative or combination therapy with Wnt pathway inhibitors, such as porcupine and/or tankyrase inhibitors, is a strategy that could halt growth of RSPO-dependent colon and breast cancers (Watson et al., 2013).
Footnotes
Authors' Contributions
C.B.C. and G.L.V.-R. designed, performed experiments, and prepared this article; M.B.B. performed experiments; S.K.R., J.E.A., and N.A.T. performed bioinformatics; T.K.S. and D.A.L. served as mentors, conceptualized, supervised, designed, and prepared this article.
Acknowledgments
The results published here are, in part, based upon data generated by the TCGA Research Network. This study utilized computing resources at the University of Minnesota Supercomputing Institute. The University of Minnesota Genomics Center provided Sanger sequencing and primer synthesis services.
Disclosure Statement
D.A.L. is the cofounder and co-owner of NeoClone Biotechnologies, Inc., Discovery Genomics, Inc., (recently acquired by Immusoft, Inc.), and B-MoGen Biotechnologies, Inc. He consults for Surrogen, Inc., and Genentech, Inc. is funding some of his research. The business of all these companies is unrelated to the contents of this article. Other authors have no conflict of interest to disclose.
Funding Information
This study was supported by grants to C.B.C. (T32 GM008244, F30 CA171547), G.L.V.-R. (T32 GM008244, T32 HL007741), D.A.L. (R01 CA134759, R01 CA113636), T.K.S. (5R00CA151672-03, P30-CA77598, Mezin-Koats Colon Cancer Research Fund, and ACS PF-06-282-01-MGO).
Supplementary Material
Supplementary Table S1
Supplementary Table S2
Supplementary Figure S1
Supplementary Figure S2
Supplementary Figure S3
Supplementary Figure S4
References
Supplementary Material
Please find the following supplemental material available below.
For Open Access articles published under a Creative Commons License, all supplemental material carries the same license as the article it is associated with.
For non-Open Access articles published, all supplemental material carries a non-exclusive license, and permission requests for re-use of supplemental material or any part of supplemental material shall be sent directly to the copyright owner as specified in the copyright notice associated with the article.