
Abstract
Effective and efficient efferocytosis of dead cells and associated cellular debris are critical to tissue homeostasis and healing of injured tissues. This important task was previously thought to be restricted to professional phagocytes (PPs). However, accumulating evidence has revealed another type of phagocyte, the amateur phagocyte (AP), which can also participate in efferocytosis. APs are non-myeloid progenitor/nonimmune cells that include differentiated cells (e.g., epithelial cells, fibroblasts, and endothelial cells [ECs]) and stem cells (e.g., neuronal progenitor cells and mesenchymal cells) and can be found throughout the human body. Studies have shown that APs have two prominent roles: identifying and removing dead cells presumably before PPs reach the site of injury and assisting PPs in the removal of cell corpses and the resolution of inflamed tissue. With respect to the engulfment and degradation of dead cells, APs are slower and less efficient than PPs. However, APs are fundamental to preventing the spread of inflammation over a large area. In this review, we present the diversity and characteristics of healthy and non-neoplastic APs in mammals. We also propose a hypothetical mechanism of the efferocytosis of immunoglobulin G (IgG)-opsonized myelin debris by ECs (APs). Furthermore, the ingestion and clearance of dead cells can induce proinflammatory or anti-inflammatory cytokine production, endothelial activation, and cellular fate transition, which contribute to the progression of disease. An understanding of the role of APs is necessary to develop effective intervention strategies, including potential molecular targets for clinical diagnosis and drug development, for inflammation-related diseases.
Introduction
Cell death occurs naturally within the human body, and the removal of dead cells is essential to maintain body homeostasis. Apoptosis is a regulated cell death program and is important in organ development, particularly in shaping new structures during mammalian embryogenesis and development (Rassoulzadegan et al., 2000; Wood et al., 2000). Conditions such as infection by microorganisms, disease, or mechanical compression can cause extensive damage to the tissue and trigger cells to undergo necrosis. Necrotic cells release their intracellular content to the surrounding milieu and stimulate proinflammatory immune pathways that can damage the surrounding healthy cells and tissues. When efferocytosis is impaired, diseases such as autoimmunity, neurological disorders, or vascular-related diseases can arise (Radi et al., 2014; Nakaya et al., 2017).
Professional phagocytes (PPs), such as macrophages and dendritic cells, are considered to be key players in removing dead cells. However, at certain time points and in certain locations during the developmental process, PP recruitment to the lesion site is delayed (Monks et al., 2005; Xi et al., 2015) or absent (Stein et al., 2004). A number of studies have suggested that other cells close to the lesion site, known as nonprofessional or amateur phagocytes (APs), are also capable of performing efferocytosis (Parnaik et al., 2000; Zhenjie et al., 2011; Li et al., 2016; Nakaya et al., 2017; Zhou et al., 2019).
APs can be observed in multiple types of non-myeloid progenitor/nonimmune cells (e.g., endothelial cells [ECs]) (Faille et al., 2012; Zhou et al., 2019), epithelial cells (Sexton et al., 2001; Hoffman et al., 2013; Fornetti et al., 2016), and neuronal progenitor cells (Zhenjie et al., 2011) in mammals (Seeberg et al., 2019), and they exert efferocytic activity when needed, particularly during disease, stress, or pathogen infection (Naonaka et al., 2017). Similar to PPs, APs can also perform efferocytosis involving three sequential steps: ligand-receptor interaction, dead cell uptake, and dead cell degradation. Dead cells expose specific ligands or secrete cytokines that attract APs, favor their uptake, and promote tissue repair, depending on their mode of death. Different types of cell death can confer proinflammatory (Tripathi et al., 2019) or anti-inflammatory (Berda-Haddad et al., 2011) signaling activity following efferocytosis.
In this review, we present recent advances in the understanding of APs, including their characteristics, the molecular mechanism of efferocytosis, and how efferocytosis affects the cellular function of APs and their surrounding environment.
Diversity and Characteristics of APs
Recently, numerous studies have documented and unveiled the existence of APs across taxa, from embryonic development to the adult stage. The role of APs in removing apoptotic or necrotic cell corpses has been most extensively studied in the invertebrate Caenorhabditis elegans. The C. elegans body possesses several APs, such as hypodermal, gonadal sheath, pharyngeal muscle, and intestinal cells. There are at least seven corpse removal genes in the C. elegans genome and they have mammalian equivalents, and are involved in signaling pathways associated with cell uptake (Henson et al., 2001). However, in this review, we mainly focus on AP diversity in mammals, particularly in healthy and non-neoplastic cells. In vertebrate embryogenesis, reactivated murine trophectoderm cells can engulf adjacent uterine epithelium debris with a notable amount of multivesicular bodies during morula blastocyst formation (Rassoulzadegan et al., 2000). Phagocytosis is also observed in macrophage-less PU.1 null mouse embryos in which mesenchymal cells act to sculpt a webbed footplate and transform the webbed regions into free interdigital spaces (Wood et al., 2000). In addition, neural progenitor cells can internalize dying neurons during adult neurogenesis (Zhenjie et al., 2011). In general, APs include different types of cells and are distributed throughout organs, including epithelia, ECs, smooth muscle cells, astrocytes, fibroblasts, Sertoli cells, and marginal reticular cells (Table 1). In addition, APs have distinct characteristics compared to PPs, which are shown in Table 2.
Diversity and Distribution of Amateur Phagocyte in Human Body
AP, amateur phagocyte.
Comparison Between Professional Phagocytes and Amateur Phagocytes
PPs, professional phagocytes.
APs can become “primary phagocytes,” which can be observed, for instance, during mammary gland involution, a process in which milk-producing epithelial cells undergo apoptosis and their population is replaced by adipose tissue. In the first 72 h, viable neighboring epithelial cells (AP) engulf apoptotic epithelial cells, while only a few populations of interstitial macrophages, which do not seem to change in number or distribution during the initial phase of the involution process, are observed outside the alveolar basement membrane. On day 6 of involution, the macrophage (M2) population increases significantly. It has been suggested that extracellular components can contribute to attracting macrophages into the area of involution. A temporal analysis revealed that collagen concentration increased as early as day 4 of involution (Monks et al., 2005, 2008; O'Brien et al., 2010). Furthermore, in spinal cord injury, PP recruitment to the site of injury is delayed up to 1 week (Xi et al., 2015), whereas microvascular endothelial cells (MVECs) act as macrophages to engulf myelin debris at the early stage of injury (Zhou et al., 2019).
APs can also become “secondary phagocytes” during the efferocytosis of dying neurons by astrocytes (APs) and microglia (PPs) in vivo (Morizawa et al., 2017; Wakida et al., 2018). The locations of microglia- and astrocyte-mediated efferocytosis are territorially segregated in vivo (Morizawa et al., 2017; Damisah et al., 2020). Microglia are found within the core of the inflamed area, while astrocytes predominantly surround the core of the inflamed area. In terms of the timing of efferocytosis initiation, microglia immediately sense inflammatory signals and begin cellular uptake at the early stage of inflammation, while astrocytes show many phagocytic inclusions at 3 days after inflammation. Astrocytes are immobile and internalize small diffuse apoptotic neuron fragments, whereas microglia migrate to injured neurons and ingest dendritic branches, which are larger than neuron fragments. This indicates that intercommunication occurs between microglia and astrocytes and affects engulfment preferences for dead cells of different sizes. A similar phenomenon was also observed in another study showing that macrophages (PPs) secrete insulin growth factor-1 (IGF-1), which binds to its receptor on epithelial cells (APs) and induces epithelial cells to engulf smaller particles and suppresses the uptake of larger particles (Han et al., 2016).
Furthermore, astrocytes have a slower kinetic rate and a lower capacity for efferocytosis than microglia (Morizawa et al., 2017; Wakida et al., 2018). The rapid clearance of debris by microglia might be associated with preventing dying cells from undergoing secondary cell death, which could worsen inflammation. Meanwhile, astrocytes remove the remaining dead cell fragments to curb the spread of inflammation and limit immune activation (Lööv et al., 2012; Morizawa et al., 2017; Wakida et al., 2018). Alternatively, there could be specific ligands on dead cells that are more favorable to detection by either microglia or astrocytes and subsequently cause the differential responses. A study demonstrated that fibroblast cells (APs) ingest late apoptotic cells more rapidly than early apoptotic cells (Parnaik et al., 2000).
Mechanism of Dead Cell Clearance by APs
Efferocytosis is defined as the engulfment and removal of cell corpses by PPs and APs. Efferocytosis comprises three sequential processes: ligand-receptor interaction, dead cell uptake, and dead cell degradation. The recognition and engagement of “eat-me” signals released by dead cells initiate dead cell internalization, through phagocytosis, clathrin-mediated endocytosis (CME), caveolae-mediated endocytosis (CvME), pinocytosis, or light chain 3 (LC3)-associated phagocytosis (LAP). The dead cells are enclosed within membrane vesicles and transported to lysosomes for degradation.
Ligand-receptor interaction
The origin of cellular debris derived from certain cell death pathways contributes to the variety of identifying signals to be recognized by phagocytes. APs possess various ligands (“eat-me” signals) and receptors (“find-me” signals) (Fig. 1 and Table 3). “Eat-me” signals are classified into two major categories: membrane-anchored and soluble bridging molecules.

Engulfment and degradation mechanism of cellular debris by amateur phagocyte. ① Engulfment of dead cell is initiated by probing specific ligand receptor. ② Ligand-receptor interaction stimulates actin polymerization and causes membrane protrusion to internalize dead cell. ③ Plasma membrane undergoes invagination to capture the dead cell and seal the dead cell in an endocytic vesicle. Endocytic vesicles are immediately targeted to the early endosome. Rab5 controls entry of the cargo from plasma membrane to the early endosome and production of PI3P. PI3P and Rab5 GTPase recruit EEA1 to regulate membrane fusion. ④ Rab7 controls progression of early endosome to late endosome and transport to lysosome. ⑤ Late endosome can fuse with lysosome and form endolysosome or phagolysosome to degrade dead cells. ⑥ Dead cells are broken down by lysozyme secreted by lysosome. EEA1, early endosome antigen 1. Color images are available online.
Types of Ligand-Receptor Pairs Within Different Amateur Phagocytes
—, Not mentioned; AIM, apoptosis inhibitor of macrophage; BAI1, brain-specific angiogenesis inhibitor 1; CD, cluster of differentiation; HCAECs, human coronary artery endothelial cells; HDM, house dust mite; HUVECs, human umbilical vein endothelial cells; IgG, immunoglobulin G; KIM1, kidney injury molecule 1; MFG-E8, milk fat globule-epidermal growth factor 8; MVECs, microvascular endothelial cells; PS, phosphatidylserine; PSR, PS receptor.
Phosphatidylserine (PS) is a membrane-anchored “eat-me” signal that is distributed in the cytoplasmic leaflet of healthy cells and is controlled by flippases, such as P4-ATPases. In dying and early apoptotic cells, PS is translocated to the cell surface to act as a signal for cell engulfment by inducing Ca2+-dependent scramblases and caspases such as transmembrane protein 16F (TMEM16F) and Xk-related protein 8 (Xkr8), respectively (Segawa and Nagata, 2015).
The binding of externalized PS to its receptor facilitates dead cell internalization. In particular, PS is mediated either directly by one or more PS recognition receptors, for example, integrin, brain-specific angiogenesis inhibitor 1 (BAI1) (Park et al., 2007), cluster of differentiation 36 (CD36), and kidney injury molecule 1 (KIM1) (Yang et al., 2015), or indirectly by soluble bridging molecules that bind PS on apoptotic cells and a receptor on phagocytes, such as milk fat globule-epidermal growth factor 8 (MFG-E8) (Segawa and Nagata, 2015). It is noteworthy that externalized PS is not completely absent from living cells; however, a certain threshold of PS exposure is required to start engulfment (Borisenko et al., 2003). Initiation of apoptosis decreases “don't eat me” signals, such as CD47 (Gerlach et al., 2020), which can further favor the uptake of apoptotic cells.
Unlike the ligand-receptor interaction mentioned above, myelin debris engulfment in vitro was shown to be facilitated by serum present in the cell culture medium (Zhou et al., 2019). This study showed that components of the serum contributed to phagocytic activity (Tanaka et al., 2001), and indeed, it has been found that natural IgG present in the serum mediates myelin debris internalization and binds to its receptor, possibly the Fcγ receptor. The Fcγ receptor is a member of a large family of proteins that consists of two major groups: classical membrane-bound surface receptors (FcγRI, FcγRII, and FcγRIII) and atypical intracellular receptors and cytoplasmic glycoproteins (neonatal Fc receptor neonatal [FcRn]). The first group is commonly expressed in myeloid cells and is not limited to the ECs and smooth muscle cells that express the FcγRII receptor (Blumberg et al., 2019).
The second group, FcRn, is found almost ubiquitously in diverse tissues throughout the human body with high expression in epithelial cells and ECs (Pyzik et al., 2019). FcRn expression is thought to be limited in newborns, but to persist throughout life. FcRn has been implicated in the transport of IgG from mother to offspring through transcytosis, thereby providing a naive and immature immune system to newborns (Pyzik et al., 2019). IgG transcytosis can also be observed in human female genital epithelial cells as a mechanism of immune surveillance (Zili et al., 2011).
Another function of FcRn is to rescue IgG from intracellular degradation through an intracellular recycling pathway in a pH-dependent manner (Goebl et al., 2008). FcRn is mostly distributed intracellularly in early or late endosomes, and only ∼7% can be found on the plasma membrane (Goebl et al., 2008; D'Hooghe et al., 2017). The acidic environment in endosomes (pH 5–6.5) is required to help FcRn bind to IgG (Goebl et al., 2008). IgG-opsonized dead cells are entrapped in pinocytotic vesicles and subsequently bind to FcRn in early endosome antigen 1 (EEA1)-, Rab5-, Rab4-, and Rab11a-positive sorting endosomes with a slightly acidic pH (∼6). Later, the IgG-bound FcRn disassembles from sorting endosomes to Rab4- and Rab11a-positive recycling endosomes and is released to the plasma membrane. Free IgG is degraded by lysosomes, while the IgG-FcRn complex fuses with Rab11a-sorting endosomes and is ready to be recycled (Pyzik et al., 2019).
Engulfment of dead cells by APs
Once ligand-receptor interactions occur, cytoskeletal rearrangement takes place to initiate the internalization process. Time-lapse video recording showed that baby hamster kidney (BHK) cells, a type of fibroblast cell line, display intermittent membrane ruffling over several hours in the cell-cell contact area, moving the apoptotic cells around on the membrane and abruptly engulfing them (Parnaik et al., 2000). In contrast, ECs and the L929 fibroblast cell line form cytoplasmic pseudopodia protrusions to probe for dead cells in the environment (Hueck et al., 2008; Lu et al., 2019). Such distinct mechanisms might be influenced by the relative proximity of dead cells to APs, as observed in astrocyte phagocytosis at the single-cell level in vitro (Wakida et al., 2018). In the membrane region, where the responding astrocytes establish physical contact with cellular debris, membrane activity (ruffling) increases and phagocytosis occurs. However, astrocytes display different behaviors when separated from debris by a greater distance: astrocytes migrate toward debris and initiate debris engulfment or, if the distance is too far, astrocytes still migrate toward debris, but cannot perform phagocytosis.
The downstream signaling mechanism resulting from ligand-receptor interactions differs depending on the engaged receptor. The engulfment of dead cells can involve protrusions of the plasma membrane that surround and engulf them into the cell. Cell membrane protrusion is tightly regulated by multiple signaling proteins. In particular, members of the Rho family of small GTPases are central to orchestrating cytoskeletal dynamics in a spatiotemporal manner (Nakaya et al., 2008). The Rho GTPase family consists of 20 members, of which Rho, Rac, and Cdc42 are the most prominent proteins involved in the migration, cell adhesion, wound healing, cell polarity, and engulfment processes. The activation of Rho GTPases is initiated by the exchange of a bound guanosine diphosphatase (GDP) nucleotide (inactive form) for guanosine triphosphatase (GTP; active form), which is mediated by guanine nucleotide factors (guanine exchange factors [GEFs]). In general, there are two types of GEF found in mammals based on sequence homology and structural features in their exchange domains: Dbl and dedicator of cytokinesis (Dock) (Spiering and Hodgson, 2011).
Among the Dock GEFs, Dock1 (also known as Dock180) is a specific activator of Rac GTPase and can bind to engulfment and cell motility protein 1 (ELMO1), forming the ELMO1-Dock1 complex, to activate Rac1 (Patel et al., 2011). Regarding how ELMO1 regulates signaling through the Dock1-Rac pathway, several different mechanisms have been reported (Lu and Ravichandran, 2006; Patel et al., 2010, 2011): the ELMO N-terminus attaches the ELMO/Dock1 complex to the plasma membrane; ELMO relieves the self-inhibitory state of Dock1 caused by the binding of SH3 to its domain; and ELMO helps Dock1 stabilize nucleotide-free Rac1 in the transition state. Activated Rac1 is able to direct localized actin polymerization to coat or grasp cargo by modulating the actin-related protein 2/3 (Arp2/3) complex near the plasma membrane by binding to and activating their effector proteins neural Wiskott-Aldrich syndrome protein (N-WASP) and Wiskott-Aldrich syndrome protein family member 2 (WAVE2). In addition, RhoG is able to bind to ELMO directly in a GTP-dependent manner and form a ternary complex (ELMO-RhoG-Dock) (Katoh and Negishi, 2003).
Rac1 activity is essential to form membrane protrusions by inducing local actin rearrangements for dead cell internalization (Marston et al., 2019). Furthermore, ELMO also recruits the actin and microtubule binding actin cross-linking family 7 (ACF7) protein to maintain directional persistence in membrane protrusion (Margaron et al., 2013). Another ELMO binding protein, Argef16, also promotes Rac1 activation in a RhoG-dependent and Dock1-independent manner in the cellular uptake mechanism (Lee et al., 2014).
Phagocytosis removes cellular debris, dead cells, and foreign particles. Phagocytosis can be performed by both PPs and APs. To perform phagocytosis, cells must undergo changes in shape through extensive membrane and cytoskeleton remodeling. One of the factors that can influence cytoskeletal remodeling is substrate stiffness. Cells are attached to the extracellular matrix (ECM), which provides structural and metabolic support. ECM composition is dynamic due to matrix deposition, cross-linking of existing matrix, and pathological conditions, which can affect its compliance (Vania et al., 2020).
Cells have the ability to sense substrate rigidity through integrin and transmit the signal to the actin cytoskeleton. Substrate rigidity can also induce cytoskeletal remodeling in cells. On soft substrates, actin shows viscous behavior, with radial flow favoring a circular morphology. As the substrate stiffness increases, actin is assembled into large, stable contractile bundles, and the membrane is spread out. These contractile bundles remodel themselves and align in a single preferential direction (Gupta et al., 2015).
As phagocytosis requires cytoskeletal remodeling, there might be a link between actin and substrate stiffness and cellular uptake activity. A small number of studies have demonstrated that the number of nanoparticles internalized on soft substrates is lower than that on stiff substrates (Huang et al., 2013; Wang et al., 2017; Xiao et al., 2019). Importantly, these studies (Wang et al., 2017; Xiao et al., 2019) showed that actin is not the key factor in stiffness-related cellular uptake. The clathrin-mediated and caveolae-mediated endocytic pathways were suggested as possible routes of particle internalization. Substrate stiffness regulates the expression of clathrin and caveolin; both proteins are more highly produced on stiff substrates than on soft substrates. To the best of our knowledge, no studies have attempted to investigate the effects of substrate rigidity on the engulfment of cellular debris or dead cells. It is unclear why actin does not play a role in nanoparticle engulfment. Is it because of the size of nanoparticles and not the substrate stiffness itself that the cell prefers to engulf them through CME and CvME? Further investigations should be performed to dissect the relationship among actin, substrate stiffness, and cellular uptake.
The shear stress imparted by interstitial flow could influence the receptor-ligand binding process. Indeed, it has recently been shown that phagocytosis of Leishmania parasites is hindered by the presence of flow (O'Keeffe et al., 2019). While the precise mechanism has yet to be elucidated, it seems highly plausible that a combination of the forces imparted by flow and the spatiotemporal gradients of chemicals and nutrients supplied by flow is likely to be a key determinant of whether a cell will engulf material. Thus, the cell stiffness and specific flow environment experienced by cells may explain the differential roles of APs depending on their location within the body.
Clearance of dead cells by APs
Internalization of dead cells relies on coordinated actin polymerization and the protrusion or ruffling of cellular membranes. The accumulation of actin on the membrane continues, forming phagocytic cups surrounding dead cells. During internalization, dead cells are wrapped within a sealed phagosome, marked by the disassembly of actin upon closure, and soon fuse with early endosomes to acquire the small GTPase Rab5 (Kim et al., 2018).
The maturation of phagosomes begins with the exchange of Rab5 to Rab7, which is a prerequisite for further phagosome maturation. In this stage, phagosomes also acquire lysosomal-associated membrane protein (LAMP), which is required for phagolysosomal fusion. In addition, the phagosome membrane acquires v-ATPase to create an acidic environment within the phagosome. Of note, the process of phagosomal acidification occurs first and is followed by phagolysosome formation (Toei et al., 2010; Dikic and Elazar, 2018).
In addition to phagosomal machinery, dead cell engulfment is mediated by IgG; dead cells are enclosed within autophagosomes, the hallmark of autophagy (Zhou et al., 2019). Autophagy is a major intracellular degradation pathway by which cytoplasmic cargo, damaged organelles, or infectious pathogens are degraded after being transported into the lysosome. Autophagy is regulated by autophagy-related genes (ATGs) and can be described in three sequential processes: autophagosome formation (phagophore formation, elongation, and maturation), autophagosome-lysosome fusion, and degradation (Hurley and Young, 2017).
The initial step of autophagy is the formation of phagophores, in which dead cells are entrapped. Phagophores require lipids and proteins to mature into autophagosomes. Recent data demonstrate that the lipid supply to build phagophores comes from the endoplasmic reticulum, mitochondria, plasma membrane, or Golgi apparatus. Generally, the formation of phagophores requires the phosphoinositide 3-kinase (PI3K) vacuolar protein sorting 34 (Vps34) along with other proteins, such as Beclin-1/ATG6, ATG14/ATG14L, and PIK3R4/Vps15. The activity of this complex is dependent on upstream autophagy regulators, including the mammalian Atg1 orthologs Unc-51-like kinase 1 (ULK1), ULK2, ATG13, and RB1-inducible coiled-coil protein 1 (RB1CC1) or FAK family-interacting protein of 200 kDa (FIP200) (Hurley and Young, 2017).
The elongation of phagophores that evolve into autophagosomes employs the E1-like ATG7 and E2-like ATG10 enzymes to conjugate the ubiquitin-like ATG12 protein to ATG5 (ATG12-ATG5). This conjugate forms a complex with ATG16L, which functions as an E3 ligase and forms the ATG12-ATG5-ATG16L complex. The second reaction involves phosphatidylethanolamine (PE) interacting with members of the ATG8 family, which comprises three subfamilies: microtubule-associated protein 1 light chain 3 (MAPL1LC3/LC3) protein, gamma-aminobutyric acid type A receptor-associated protein (GABARAP), and Golgi-associated ATPase enhancer of 16 kDa (GATE16). LC3-I is produced by proteolytic cleavage of pro-LC3 or ATG8 mediated by ATG4 (a cysteine protease), which leaves an exposed C-terminal glycine that is amenable to conjugation by ATG7. ATG3 then conjugates LC3-I to PE at the carboxy terminal glycine to form LC3-II. LC3-II is then brought into the membrane to continue extending the phagophore and ultimately form autophagosomes. LC3-II is distributed along the membrane and helps select the cargo to enter the autophagosome and fuse with the membrane (Dikic and Elazar, 2018).
As the phagophore continues to elongate, autophagosomes undergo maturation, which involves ATG8 and PI3K dissociation by ATG4 from the nascent autophagosome outer membrane, recruitment of machinery responsible for lysosomal delivery, and fusion with lysosomes. The autophagosome fuses with the lysosome to begin the process of content degradation, which is mediated by soluble N-ethylmaleimide-sensitive factor attachment protein receptor (SNARE). ATG14 binds to the autophagosomal outer membrane by interacting with syntaxin 17 (STX17) and modulates membrane tethering to enhance SNARE-mediated fusion. The degraded products are recycled to the cytosol through permeases. PE is removed from LC3-II in the cytoplasm by ATG-4, which allows LC3-I to be recycled (Dikic and Elazar, 2018).
Proposed model of IgG-opsonized myelin debris in ECs (AP)
Zhou et al. (2019) found that IgG facilitated myelin debris internalization by ECs and that autophagy machinery was activated to degrade myelin debris. Based on RNA sequencing data from Zhou et al. (2019), we selected several genes associated with trafficking routes, such as LAP, CME, and CvME, and created a heat map to illustrate their gene expression (Fig. 2). Furthermore, from the RNA sequencing data, we proposed a mechanism for the efferocytosis of IgG-opsonized myelin debris by ECs (Fig. 3).
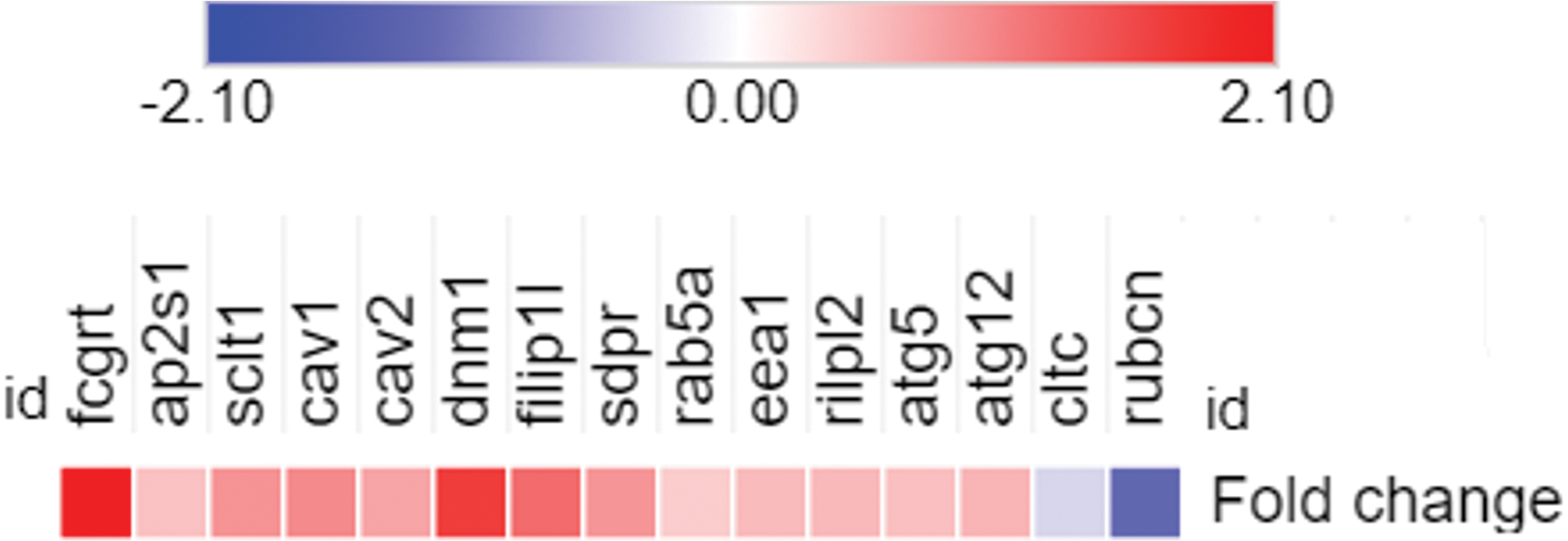
Heatmap gene expression of selected genes related to LAP, CME, and CvME, which is based on RNA sequencing data from Zhou et al. (2019). CME, clathrin-mediated endocytosis; CvME, caveolae-mediated endocytosis; LAP, LC3-associated phagocytosis; LC3, light chain 3. Color images are available online.
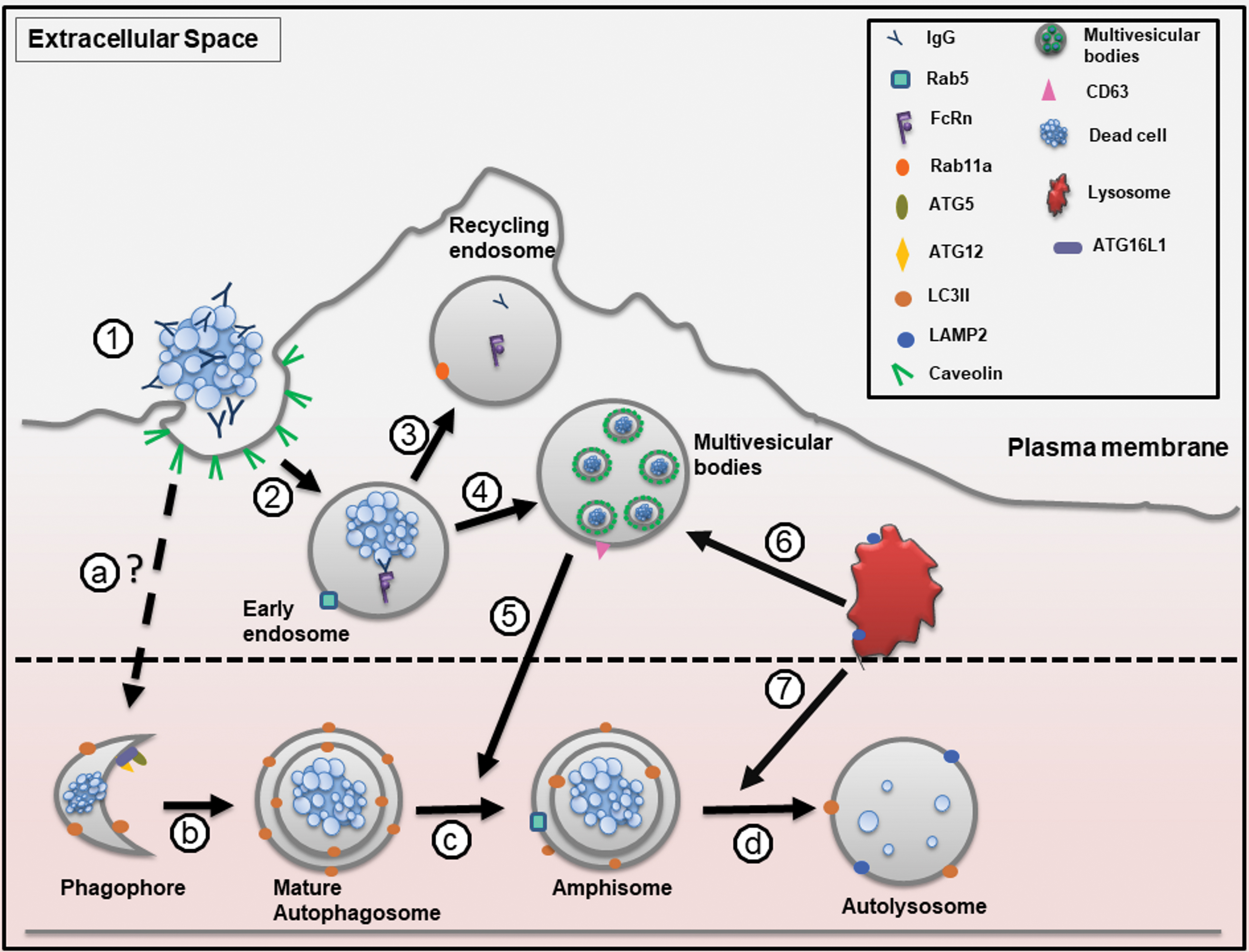
Proposed model of IgG-opsonized myelin debris in endothelial cell. ① Myelin debris is opsonized by IgG. IgG-opsonized myelin debris penetrates into amateur phagocyte by caveolar transcytosis. ② IgG-myelin debris and FcRn are engaged within the early endosome. ③ IgG is shipped back to the extracellular space through exocytic vesicle for recycling purpose. ④ Early endosome-contained myelin debris proceeds to multivesicular bodies. ⑤ Multivesicular bodies can fuse with autophagosome to form amphisome. ⑥ Lysosome can fuse with multivesicular bodies or ⑦ amphisome to begin degradation Autophagy is the main route for myelin debris degradation: (a) dead cells are opsonized by IgG. IgG-opsonized dead cell crosses the plasma membrane through clathrin-mediated transcytosis. IgG-opsonized dead cells are entrapped within endocytic vesicle. However, it is unclear how IgG-myelin debris can enter the autophagy process. ATG12-ATG15 complex interacts with ATG16L1 to determine the sites of autophagosome synthesis by regulating the targeting of LC3 to ATG5-ATG12-associated membranes. LC3II binds to the nascent phagophore and helps to elongate the phagophore to form autophagosome. (b) Double-membrane autophagosome is fully matured and ATG5-ATG12-ATG16L1 complex is dissociated from plasma membrane and LC3II remains on the autophagosome. (c) Early endosome (Rab5) or multivesicular bodies can fuse with autophagosome to form amphisome. (d) Amphisome fuses with lysosome to degrade the cargo. ATG, autophagy-related gene; FcRn, Fc receptor neonatal; IgG, immunoglobulin G. Color images are available online.
LAP is stimulated upon the phagocytosis of dead cells, which that engages innate immune receptors (e.g., Toll-like receptor [TLR], T cell immunoglobulin and mucin domain containing 4 [TIM4], and Fcγ receptor). The ligand-receptor interaction causes the recruitment of LC3 (ATG8) to the phagosome membrane to form a LAPsome. LAP occurs in phagocytic cells such as macrophages, ECs, and epithelial cells. LAP proceeds independent of the autophagic preinitiation complex containing ATG13, ULK1, and FIP2000, but requires autophagic machinery such as Beclin1, PI3P, ATG5, and ATG7. In the early stages of LAPsome formation, the RUN domain-containing cysteine-rich protein (Rubicon) is required to generate PI3P from a complex that contains Beclin1, ultraviolet radiation resistance-associated gene protein (UVRAG), and Vps34. Rubicon negatively regulates autophagy by disrupting autophagosome-lysosome fusion (Herb et al., 2019). Zhou et al. (2019) found that the expression of the rubcn gene (which encodes Rubicon) was downregulated, indicating that LAP was inactive. Another hallmark of LAP is the single-layer membrane of phagosomes, which is absent during myelin debris degradation. In fact, Zhou et al. (2019) found several double-layer membrane structures called autophagosomes, which are distinct features of autophagy. Therefore, this evidence emphasizes that LAP is not the pathway used to degrade IgG-opsonized myelin debris.
CME is a vesicular transport event that facilitates the entry of a wide range of extracellular molecules into the cell and recycles receptors. CME is characterized by two major protein categories, clathrin and adaptor proteins. Clathrin is a triskelion structure composed of three light chains (CLTA) and three heavy chains (CLTC). Initiation of CME requires the binding of phosphatidylinositol-4,5-bisphosphate (PIP2) and APs, such as AP2, at the cytoplasmic face of the plasma membrane. Then, clathrin binds to AP2 and assembles to form a coat around the vesicle (Kaksonen and Roux, 2018). RNA sequencing data indicated that two clathrin-associated genes, ap2s1 and sclt1, are upregulated during myelin debris engulfment, indicating that CME might be used to engulf myelin debris. However, the expression of the cltc gene, which encodes clathrin heavy chain (CLTC), is downregulated. For this reason, it is unclear whether clathrin plays a role in myelin debris internalization.
Zhou et al. (2019) found that serum IgG was required for myelin debris uptake. Then they tested several receptors that commonly interact with IgG, such as complement-3 receptor (CR3), Mac-2 (Galactin-3), and low-density lipoprotein receptor-related protein 1 (LRP1). However, none of these receptors facilitated myelin debris engulfment. The RNA sequencing data showed that the fcgrt gene, which encodes the FcRn receptor, was upregulated, suggesting a possible interaction between IgG and FcRn through transcytosis. Transcytosis is a mechanism of transcellular transport of particles or molecules from one side of the cell to the other side through discrete plasmalemmal vesicles or caveolae.
Interestingly, the expression of genes related to caveolae, such as cav-1 (caveolin-1), cav-2 (caveolin-2), filip1l, dnm1 (dynamin1), and sdpr (serum deprivation-response protein)/cavin2, is increased, suggesting that caveolar transcytosis might be an alternative route for myelin debris uptake. Caveolar transcytosis involves tightly controlled steps, including caveolar budding at and scission from the apical membrane to form transport vesicles. Caveolar budding and scission rely on phosphorylated caveolin as well as dynamin for pinching off from the plasma membrane (Simmons et al., 2018). It is postulated that within the early endosomal compartment, which is marked with EEA1, the IgG-myelin debris complex binds to the FcRn receptor under acidic conditions. IgG is disassembled from FcRn and returned to the extracellular space through exocytic vesicles marked by the Rab11 protein (Pyzik et al., 2019). Of note, endocytosis (CvME) is not the main route for the transportation of myelin debris to lysosomes, as indicated by the minimal colocalization between myelin debris and early or late endosomes.
Zhou et al. (2019) found that autophagy was activated to degrade myelin debris; nevertheless, it is still unclear how caveolin integrates with autophagic machinery. The RNA sequencing data show two upregulated genes, namely atg5 and atg12, which are commonly found in phagophores. Phagophores are characterized by the ATG5-ATG12-ATG16L1 complex, in which ATG16L1 recruits the LC3II synthesis complex (ATG12-ATG5 complex) to the nascent phagophore. The autophagosome is fully mature when the ATG5-ATG12-ATG16L1 complex is dissociated from the plasma membrane and only LC3II is left on the plasma membrane (Ravikumar et al., 2010; Qian et al., 2017).
Autophagosomes can also fuse with early or late endosomes to form amphisomes. Later, amphisomes fuse with lysosomes to form autolysosomes. Genes related to proton transfer, such as Atp6v0e, Atp6v1e1, Atp6v1h, and Atp6v1d, are activated to mediate lysosome acidification for dead cell degradation (Toei et al., 2010; Zhou et al., 2019). The LAMP2 protein is present in lysosomes to facilitate the fusion of autophagosomes with lysosomes and transport digestive enzymes and cellular materials into autophagosomes (Dikic and Elazar, 2018; Zhou et al., 2019).
The Different Sources and Characteristics of Dead Cells in APs
Cell death is an important process in the body, as it promotes the removal of unwanted cells. There are three major cell death programs that have distinct features: apoptosis, necrosis, and autophagy (described in Table 4) (Inbal et al., 2002; Okada et al., 2004). Apoptosis is controlled cell death that activates initiator caspase proteins, which are dormant in the normal physiological state, leading to cleavage-dependent activation of effector caspase proteins. This, in turn, promotes nuclear and cytoskeletal fragmentation and the activation of accessory enzymes, such as caspase-activated DNase (CAD), to degrade chromosomal DNA. Proteolysis of actin, fodrin, and gelsolin causes cell shrinkage and membrane blebbing to form apoptotic bodies (AptBs). Apoptosis prevents the cell content from leaking out to the extracellular environment, therefore inhibiting immune activation and allowing the components to be recycled.
Comparison of Apoptosis, Necrosis, and Autophagy
+, Present; −, absent; Bcl2, B cell lymphoma 2; DAP, death-associated protein; mTOR, mammalian target of rapamycin; PARP, poly (ADP-ribose) polymerase; PI3K, phosphoinositide 3-kinase.
Apoptotic cells include two subtypes: large membrane-bound vesicles and smaller apoptotic microparticles. Apoptotic cells also release a large number of 1–5 μm extracellular vesicles named AptBs that can be identified and engulfed by phagocytes for clearance purposes (Atkin-Smith et al., 2015). In particular, apoptosis also generates 0.1–1 μm microparticles that lack nuclei or synthetic capacity; these microparticles may contain cytoskeletal proteins and have some quantity of PS (Hargett and Bauer, 2013). Their composition and functional properties vary with their cellular origin and the type of stimulus involved in their formation.
Necrotic cell death can be stimulated by injury, infection, cancer, oxygen depletion, or toxins. Typically, necrotic cells lack cell membrane integrity, and their contents leak into the extracellular environment, which later attracts PPs to initiate a proinflammatory response. Necrotic cells produce cell remnants, cellular debris, or microparticles as a result of plasma membrane rupture. Interestingly, studies have prompted reconsideration of necrosis as multiple distinct types of events, at least some of which are tightly regulated and not always accidental. Necroptosis is a form of regulated necrotic cell death that arises in response to stress stimuli such as interferons, death ligands, or TLR activation. It shares similar pathways with apoptosis, for example, the inhibition of caspase-8 (apoptosis) activates the receptor interacting protein kinase 1/3 (RIP1/3) complex (necrosis) through phosphorylation and mediates necroptosis. Phosphorylated RIP3 leads to the recruitment and phosphorylation of mixed lineage kinase domain-like (MLKL), which cause membrane damage and necroptosis (Chen et al., 2018).
Unlike apoptosis and necrosis, autophagy is a natural process of degrading cellular contents during nutrient stress. It can be distinguished by the presence of double membrane vesicles, autophagosomes, which fuse with lysosomes to form autolysosomes. This event begins with mammalian target of rapamycin (mTOR) and ATG proteins (Chen et al., 2018). Autophagy is also involved in cell death, which has been observed in adult hippocampal neural cells in response to insulin withdrawal. Autophagy has distinct features, including extensive autophagic vacuolization of the cytoplasm with no changes in chromatin organization, which are seen in apoptosis (Ha et al., 2019).
Microparticles are a heterogeneous group of vesicles (100–1000 nm) originating from blood or cells following chemical or physical activation, necrosis, or apoptosis. Microparticles carry bioactive molecules (originating from the plasma membrane, cytosolic fraction, cytoskeleton, mitochondria, or DNA) derived from their parental cells. The structure, size, and dimension of microparticles depend on the cell-activating stimulus. For instance, in an in vitro study, thrombin was able to induce deep invagination of the plasma membrane of red blood cells, leading to the formation of knobs and blebs on its surface and causing platelet disintegration into fragments (Ponomareva et al., 2017).
A high rate of cell death is an inevitable consequence of impaired cell function due to external stimuli (e.g., environmental pollutants, allergens, and pathogens) (Hoffman et al., 2013) or internal stimuli (e.g., autoimmune disease) (Shinde et al., 2018). Pathogen elimination by PPs can induce cell death in the PP itself (e.g., bursting of neutrophils after Leishmania major engulfment) and then clearance of the dead phagocytes by other PPs (e.g., macrophages). Interestingly, specific pathogens can direct specific cell death fates, for example, Mycobacterium tuberculosis-infected macrophages stimulate necrosis rather than apoptosis (Martin et al., 2014).
While macrophages are able to identify and engulf cells during the early stages of apoptosis, APs such as fibroblasts and lens epithelial cells can effectively engulf preaged apoptotic cells (Parnaik et al., 2000). The nature of this phenomenon is unclear; however, it is assumed that time is a critical factor for apoptotic cells to acquire specific features that only appear late in the death process, probably including the concentration of the “find me” signal, for internalization by APs. AptBs, necrotic cells, and microparticles can be engulfed and eliminated by APs. Furthermore, the type of cell death also affects the preference of AP engulfment, as Lu et al. (2019) found that necroptotic and pyroptotic cells were engulfed by APs more quickly than apoptotic cells. Necroptotic cell uptake by PPs was inefficient due to the expression of the “don't eat me” signal, CD47 (Gerlach et al., 2020), whereas pyroptotic cells were internalized by PPs more efficiently than heat-killed necrotic cells (Wang et al., 2013). It is still unclear how APs manage to recognize and internalize necroptotic cells.
The Clinical Implications of Phagocytosis by APs
Ingestion by PPs or APs is the fate of most cells that undergo cell death. Ingestion can potentially exacerbate inflammation due to proinflammatory cytokines and leukocyte infiltration. In addition, dead cells might trigger cellular fate transition in APs, which may lead to fibrotic disease. Conversely, several studies have found conflicting results demonstrating that dead cells confer advantages to APs, such as antiapoptotic and anti-inflammatory effects.
Inflammatory response
Generally, the uptake of apoptotic cells produces anti-inflammatory responses (transforming growth factor beta [TGF-β] and interleukin [IL]-10) and decreases the secretion of proinflammatory cytokines (tumor necrosis factor alpha [TNF-α], IL-1β, and IL-8). Phagocytosis of necrotic cells commonly leads to proinflammatory signaling due to the leakage of cellular material into the extracellular environment. However, APs secrete two different types of cytokines after engulfing AptBs: (1) anti-inflammatory cytokines and (2) proinflammatory cytokines, which activate ECs and promote leukocyte sequestration, thus modulating the inflammatory response. Various cytokines released by APs after cellular debris engulfment and degradation are summarized in Table 5.
Cytokine Releases After Cell Engulfment by Amateur Phagocytes
—, not mentioned; BMECs, brain microvascular endothelial cells; HAECs, human aortic endothelial cells; IL, interleukin; I/R, ischemia/reperfusion; MCP1, monocyte chemoattractant protein 1; MIP2, macrophage inflammatory protein 2; MLECs, murine lung endothelial cells; TGF-β, transforming growth factor beta; TNF, tumor necrosis factor; VEGFi, vascular endothelial growth factor inhibitor; WT, Wild Type.
Phagocytic clearance of apoptotic cells by kidney epithelial cells subsequently downregulated nuclear factor kappa-light-chain-enhancer of activated B cell (NF-κB) activity, resulting in an anti-inflammatory phenotype in proximal tubular cells that included reduced TLR4 expression and proinflammatory cytokine production and a decreased ability to activate macrophages (Liu et al., 2018). Moreover, apoptotic Jurkat cell engulfment by LR73 cells and peritoneal macrophages led to similar transcriptional changes, such as decreased expression of proinflammatory genes and increased expression of actin rearrangement/cell motility and anti-inflammatory genes (Morioka et al., 2018). Airway epithelial cells generated anti-inflammatory cytokines, such as TGF-β and prostaglandin E2 (PGE2), in a PS- and Rac1-dependent manner after apoptotic cell treatment Rac1 (Juncadella et al., 2013).
In contrast, a recent study showed that engulfment of myelin debris by MVECs stimulates a proinflammatory response by inducing EC angiogenesis (Zhou et al., 2019). Similarly, the engulfment of apoptotic ECs by human MVECs results in an increased expression of proinflammatory chemokines and enhanced binding of leukocytes to human MVECs (Kirsch et al., 2007). Circulating cellular debris in the blood promotes the attachment of platelets due to von Willebrand factor (vWF) expression on the EC surface and the involvement of glycoprotein Ib and P-selectin. Debris is then internalized by ECs and activates reactive oxygen species synthesis through the xanthine/xanthine oxidase system and nicotinamide adenine dinucleotide phosphate (NADPH) oxidase (Terrisse et al., 2010).
The discrepancy between the two different results (anti-inflammatory vs. proinflammatory responses) is probably due to differences in apoptotic cell sources or activation mechanisms. The composition of AptBs and cellular debris is highly related to their cellular origin and to the type of stimulus involved in their formation. Therefore, the cellular events caused by their engulfment by APs may also be different. Apoptotic human umbilical vein endothelial cells have two different forms: microparticles (described as <1 μm, AnnexinV+/DAPI−/histone−) and AptBs (1–3 μm, AnnexinV+/DAPI+/histone+). AptBs contain IL-1α and are able to induce the chemokines IL-8 and monocyte chemoattractant protein 1 (MCP1), whereas microparticles lack IL-1α and are thus incapable of inducing proinflammatory chemokines (Berda-Haddad et al., 2011).
In addition, the inflammatory response is also affected by the organelles attached to the dead cells. ECs treated with MAPK-activated protein kinase 2 (MAPKAPK2 or MK2) generated microparticles with dysfunctional mitochondria that had proinflammatory effects on ECs. Interestingly, microparticles from MK-2-deficient cells improved endothelial functions by reducing the intercellular adhesion molecule 1 (ICAM1) and E-selectin and rescued cardiac hypertrophy (Tripathi et al., 2019).
The involvement of dead cells in cell survival is still controversial. Considering the conflicting results above, features such as organelles and biomolecules attached to dead cells should be taken into consideration in the study of APs, as they play pivotal roles in AP survival.
Antiapoptotic effect
AptBs, necrotic cells, and microparticles carry bioactive molecules, such as RNA, lipids, and peptide molecules, which can affect the surrounding cells either in a paracrine or autocrine manner. Macrophages, the main players in eliminating AptBs, gain antiapoptotic benefits through the mediator sphingosine-1-phosphate (S1P) that leads to macrophage polarization to anti-inflammatory M2 macrophages (Weigert et al., 2006). S1P activates erythropoietin signaling to promote cellular debris clearance and immune tolerance (Luo et al., 2016).
Similar phenomena have also been observed in APs. In response to atherosclerosis, EC-derived AptBs carry microRNA (miRNA)-126, which is then transferred to recipient cells and triggers C-X-C motif chemokine 12 (Cxcl12) production. Cxcl12 promotes progenitor cell mobilization to the injured area; thus, it creates plaque stability and acts as an antiapoptotic factor (Zernecke et al., 2009). Another example is that microparticles from ECs can protect acceptor cells from apoptosis by inhibiting p38 activity (Jansen et al., 2012). Therefore, EC-derived microparticles contribute to the sorting of several proapoptotic factors and prevent cell detachment and apoptosis. The ability to inhibit apoptosis can be analyzed from physiological changes such as the activation/upregulation of mitogenic signaling pathways (i.e., PI3K/Akt, extracellular signal-regulated kinase [Erk] 1/2), the downregulation of apoptotic proteins (Bad, BCL2-associated X [Bax], caspase 9, glycogen synthase kinase 2 [GSK2], and Forkhead box O 1 [Foxo1]), and the upregulation of a number of antiapoptotic genes (B cell lymphoma 2 [Bcl2], cellular FLICE-like inhibitory protein [cFLIP], heat shock protein [HSP] 72, and HSP90) (Dignat-George and Boulanger, 2011; Jiang et al., 2015; Westman et al., 2020).
EC activation
Dead cell engulfment can cause an inflammatory response and trigger APs to become activated in response to proinflammatory cytokines. During activation, ECs alter their morphology and function, which can be a protective response to provoking factors or could trigger inflammatory disorders and end-organ damage (Kirsch et al., 2007). Activation of ECs involves upregulated expression of several proteins, such as ICAM1, endothelial-leukocyte adhesion molecule 1 (ELAM1), and vascular endothelial adhesion molecule 1 (VCAM1). These proteins are expressed at the EC surface and are critical to facilitate leukocyte binding and transmigration across the endothelium (Chang et al., 2017; Kim et al., 2018; Zhou et al., 2019). Leukocyte binding does not occur when ECs are treated with necrotic cells (Kirsch et al., 2007). Furthermore, endothelial activation is also characterized by increased synthesis of nitric oxide (NO) to dilate blood vessels and increase vascular permeability (Tesse et al., 2005). Activation of ECs is a transient process that returns to basal conditions when proinflammatory cytokines are reduced (Pinte et al., 2016).
Cellular fate
Inflammation is postulated to be a potential trigger for cell fate transition by increasing TGF-β expression (Monks et al., 2005; Zhou et al., 2019). Inflamed cells can undergo cell death and release cellular debris that is recognized by PPs and APs. Proinflammatory cytokines such as IL-6 and IL-8 can induce fibroblast-like cells through a process resembling endothelial-to-mesenchymal transition (EndMT). miRNAs, such as miRNA125b (Ghosh et al., 2012) and miRNA126 (Zhang et al., 2013), also participate in the EndMT process.
EndMT cells exhibit an elongated morphology, increased alpha smooth muscle actin (αSMA) expression, downregulated proinflammatory cytokine production, and the expression of several proteins such as CD31, vWF, and VE-cadherin, which are molecular markers of ECs (Nakaya et al., 2017; Zhou et al., 2019). In particular, engulfment of myelin debris by brain microvascular endothelial cell induces endoMT marker α-smooth muscle actin synthesis and reduces CD31 (Zhou et al., 2019). The detailed molecular mechanism activated by TGF-β in APs upon dead cell engulfment is understudied. Other studies have shown that the EndMT process might be associated with Smad-dependent and Smad-independent pathways, as well as transcriptional regulators such as Snail1, Snail2, and Twist (Medici et al., 2011; van Meeteren and ten Dijke, 2012).
Conclusion and Future Perspectives
APs are distributed throughout the human body and include different types of cells. Even though they have distinct features, APs are able to perform PP tasks to remove dead cells and associated cellular debris from the cellular environment. Previous studies have identified and proven the pivotal role of APs during developmental processes and pathological conditions. Furthermore, PPs and APs share some similar molecular machinery to engulf and degrade dead cells. Finally, degradation of dead cells induces the secretion of inflammatory cytokines and affects cell survival and cell fate.
There is no doubt that AP research is still in its infancy, and further investigation is needed to determine how APs react to inflammation: Do APs preferentially engulf certain types of dead cells? How does dead cell engulfment benefit APs? What kind of machinery differentiates APs from PPs? Moreover, the roles of mechanical cues such as stiffness and shear stress from fluid flow deserve further attention, as it seems certain that they too play important roles in AP function. By addressing these issues, we can gain deeper insight into the role of APs in disease progression and identify regulatory mechanisms and drug targets.
Footnotes
Acknowledgments
The author would like to thank Dr. Richard Daniel and Dr. Lingjuan Wu at Newcastle University (UK) for critical reading and revising.
Disclosure Statement
The authors declare no competing interests.
Funding Information
This work was supported by the National Natural Science Foundation of China (12032007, 31971242, and 31701275), the Natural Science Foundation of Chongqing (cstc2019jcyj-zdxmX0028), and Chongqing Municipal Education Commission, China (KYYJ202001), as well as Fundamental Research Funds for Central Universities (2019CDYGZD008).