
Abstract
N1-methyladenosine (m1A) is a prevalent RNA modification widely affecting RNA structural stability, folding, and interactions with proteins. Recently, there have been increasing reports on the roles of m1A regulators in tumors. However, their mechanisms and clinical relevance remain unclear. This study systematically evaluates the epigenetic characteristics and clinical relevance of m1A regulators using bioinformatic methods. Our results show widespread gene expression changes for m1A regulators, which are related to the activation and inhibition of carcinogenic pathways and overall patient survival. Collectively, this investigation provides new insights into assessing tumor prognosis and targeted therapy.
Introduction
Gene expression is a multistep process and can be regulated at various levels, including dynamic and reversible post-transcriptional RNA modifications (Dominissini et al., 2016; Sharma et al., 2018). Technology development in recent years, such as high-throughput sequencing methods, has enabled the identification of many types of post-transcriptional RNA modifications at thousands of sites in tRNA, rRNA, messenger RNA (mRNA), snRNA, and other RNA species (Sánchez-Vásquez et al., 2018; Zhao et al., 2019). These sequencing technology breakthroughs have also greatly improved our understanding about the location, regulation, and function of RNA modifications in the transcriptome (Li et al., 2017; You et al., 2017).
More than 100 post-transcriptional modifications have been discovered in RNA, of which methylation modifications account for over half (Zhang and Jia, 2018; Shima and Igarashi, 2020; Zhao et al., 2020b). N1-methyladenosine (m1A) is one such post-transcriptional RNA modification identified decades ago in RNA from several species, including humans (Dunn, 1961; Dominissini et al., 2016; Dai et al., 2018). m1A is a common modification found in several RNA types, such as noncoding RNA and mRNA (Li et al., 2017), which enhances local structural stability and induces correct folding of the RNA (Oerum et al., 2017). In addition, the positive charge carried by m1A affects RNA interactions with proteins (Zhao et al., 2019). However, the mechanism for m1A formation and regulation has not been fully elucidated.
Similar to m6A, m1A is governed by several types of regulators, including writers, readers, and erasers (Niu et al., 2018; Chen et al., 2019a). The writers (TRMT10C, TRMT61B, TRMT6, and TRMT61A) and readers (YTHDF1, YTHDF2, YTHDF3, and YTHDC1) usually function as methyltransferases and RNA binding proteins (Engel and Chen, 2018; Zhao et al., 2020a), while the two erasers, ALKBH3 and ALKBH1, typically function as demethylases (Deepa et al., 2018; Dinescu et al., 2019). The discovery of these m1A regulators has greatly advanced our understanding of how RNA methylation functions in regulating gene expression.
It is reported that m6A regulators' dysregulation involves multiple cellular processes, such as reduced cell proliferation, impaired self-renewal, developmental defects, and cell death, and is furthermore involved in the development of different types of tumors (Safra et al., 2017; Vu et al., 2017; Delaunay and Frye, 2019; Yue et al., 2019; Liu et al., 2020b). Yang et al. (2020) found that RNA N6-methyladenosine reader IGF2BP3 regulates cell cycle and angiogenesis in colon cancer. Furthermore, downregulation of m6A reader YTHDC2 promotes tumor progression and predicts poor prognosis in nonsmall cell lung cancer (Sun et al., 2020). And recently, the relationship between m1A regulators and tumor genesis had been reported. Pi et al. (2020) found that YTHDF1 promotes gastric carcinogenesis by controlling translation of FZD7. m1A regulator TRMT10C predicts poorer survival and contributes to malignant behavior in gynecological cancers (Wang et al., 2020b). Esteve-Puig et al. (2020) implied that epigenetic loss of m1A RNA demethylase ALKBH3 in Hodgkin Lymphoma targets collagen conferring poor clinical outcome. And Zheng et al. (2020) illustrated that cytoplasmic m1A reader YTHDF3 inhibits trophoblast invasion by downregulation of m1A-methylated IGF1R. However, systematic studies about the relationship between m1A regulators and hepatocellular carcinoma are still lacking.
In this study, the molecular alterations of m1A regulators and their clinical relevance were investigated. We analyzed the clinical and sequencing data of 33 cancer types from The Cancer Genome Atlas (TCGA) and detected the genetic alterations in m1A regulators. After evaluating the regulators related to carcinogenic pathways, we then assessed the relationship between the genetic alterations and overall survival (OS), progression free interval (PFI), and disease specific survival (DSS) of cancer patients. We found a prevailing inheritance and expression changes of m1A regulators across the 33 cancer types, indicating that m1A dysregulation was important in different tumors. Furthermore, m1A regulatory protein expression was correlated with the activation or inhibition of various carcinogenic pathways, demonstrating that the writers, readers, and erasers may play key roles in tumor development and progression. The relationship between m1A regulator expression and patient OS indicated that m1A regulators have varying potential for prognostic assessment in specific types of cancer and may inform on new treatment strategies.
Materials and Methods
The dataset acquisition and process of pan-cancer
This study was approved by the ethics committees of The First Affiliated Hospital of Zhengzhou University. We identified 10 m1A regulators from recently published literature. All somatic mutations and copy number variation (CNV) data were from TCGA and the download data type was Level 3, and the 33 cancer type information is shown in Supplementary Table S1. The Genomic Identification of Significant Targets in Cancer (GISTIC 2.0) algorithm was applied to identify significant focal regions of gains and losses (Brown et al., 2020). RNA sequence data and clinical data were obtained from the TCGA database as described previously (Wang et al., 2020a).
Survival analysis related to m1A regulators
We divided all patients into two groups based on the median expression of m1A regulators to explore whether the expression of m1A regulators were related to patient survival (Pi et al., 2020). Kaplan–Meier and log-rank tests were adopted to analyze the survival time of patients. The significance level was considered as p < 0.05.
Identification of differentially expressed genes
We used DESeq to identify differentially expressed genes between tumors and paired normal tissues in each cancer type. Genes with an adjusted p < 0.01 and fold change ≥2 were identified as differentially expressed genes.
Oncogenic pathway activity across cancer types
To calculate the activity of cancer-related pathways, we first converted the transcripts per million (TPM)-based gene expression to Z-score using the zFPKM package (Hart et al., 2013; Li et al., 2019). Gene set variation analysis was performed to assess the underlying changes in pathway activity, which is a nonparametric unsupervised method that transforms the genes of the sample matrix into predefined gene sets without a priori knowledge of experiment design (Chen et al., 2020).
Protein–protein interaction analysis among m1A regulators
Protein–protein interaction (PPI) network information was gained through The Search Tool for Retrieval of Interacting Genes database (Hu et al., 2020), which is an online database analyzing functional protein association networks and providing insights into the progression or development of diseases (Franceschini et al., 2013). And those experimentally validated interactions with a combined score >0.4 were selected as significant. The screened networks were visualized by Cytoscape 3.7.0.
Statistical analysis
The Pearson correlation coefficient (PCC) was used to evaluate the correlation between two variables, including m1A regulatory protein expression and pathway activity and the correlation among the m1A regulators. PCC >0.5 and p < 0.05 were considered to be significantly related. Kaplan–Meier analysis was adopted to detect the difference between two subgroups, and log-rank p < 0.05 was considered to be significantly related.
Results
Extensive genetic alterations in m1A regulators across 33 cancer types
Based on existing literature, we identified and screened for 10 genes that function as m1A regulators, including 4 writers, 4 readers, and 2 erasers (Fig. 1A, B). The incidence of genetic changes to m1A regulators across 33 cancer types was identified by somatic mutation and CNV data. Overall, the average mutation frequency of m1A regulators ranged from 0% to 6% across 33 cancer types. On average, reader genes showed a higher mutation frequency (Fig. 2A). Next, we studied the CNV frequency for all m1A regulators and found that CNVs were common in cancer. TRMT6, YTHDF1, and YTHDF3 showed extensive CNV amplification. In contrast, CNVs in TRMT61A, ALKBH1, and ALKBH3 were generally absent (Fig. 2B).
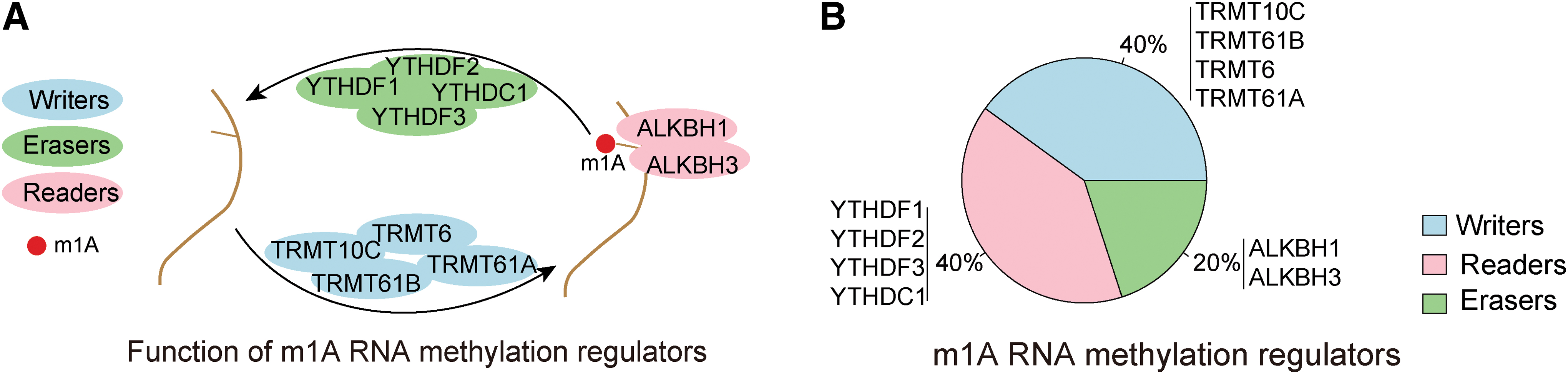
Identification and screening of m1A regulators.
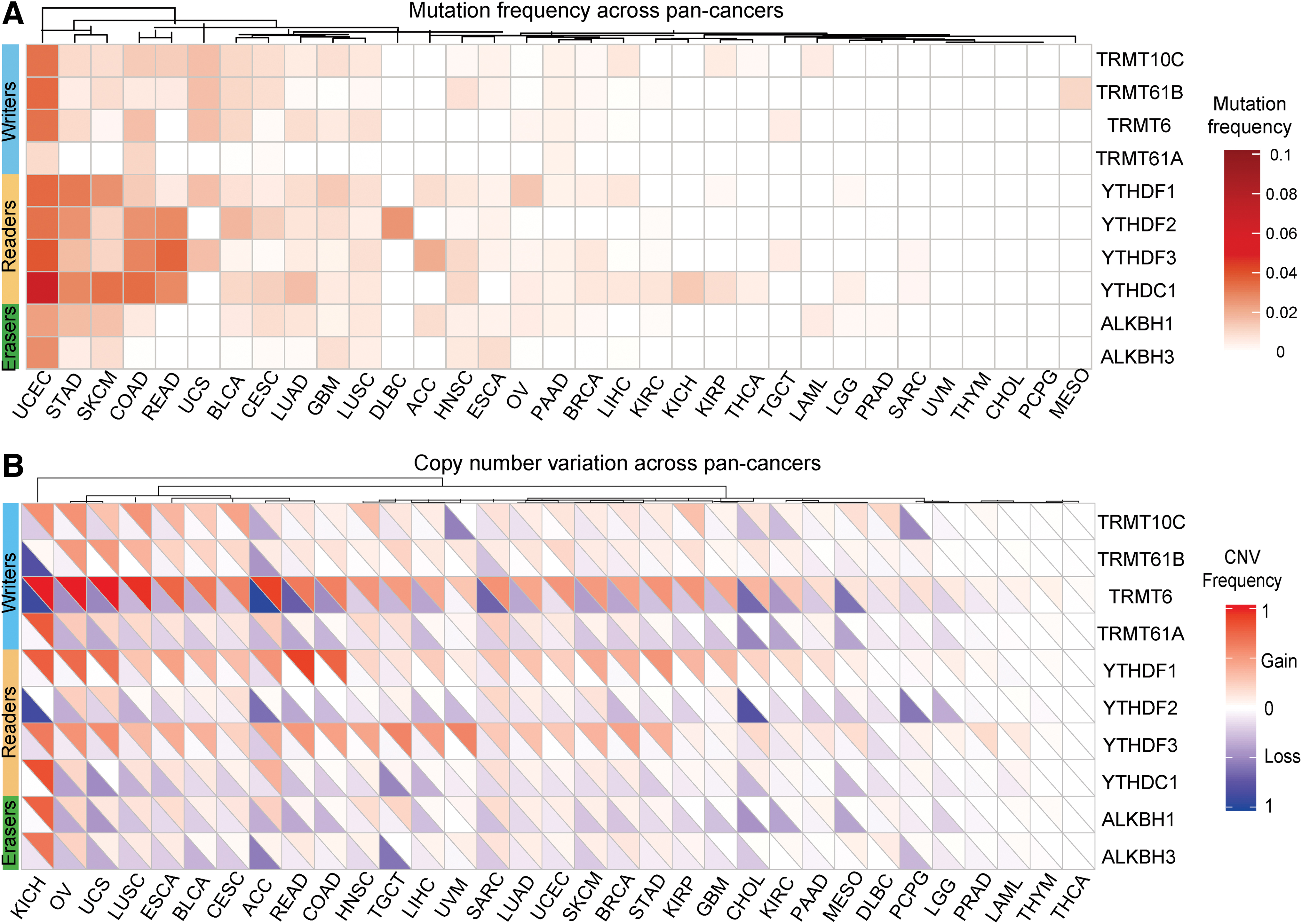
Genetic alterations of m1A regulators across cancers.
We further explored m1A regulator expression across 24 cancer types to clarify whether these genetic variations resulted in expression changes. The level of mRNA expression of all 10 m1A regulatory genes was associated significantly with CNV. Compared to normal tissue, m1A regulator expression was significantly increased in cancer with amplified CNVs, whereas the expression of m1A regulators was significantly reduced when CNVs were deleted such as in liver hepatocellular carcinoma (LIHC) and kidney chromophobe (KICH) (Fig. 3A and Supplementary Fig. S1). In particular, we found that TRMT6 expression was significantly increased across 24 cancer types (Fig. 3B). In conclusion, these results showed a high inheritance and expression changes of m1A regulators across the different cancer types.

Expression alterations of m1A regulators across cancers.
Clinical significance of m1A regulators in cancer
Genetic and expression changes found in m1A regulators across cancers can provide new insight into the development of cancer treatment. We next focused on associations between m1A regulators and patient OS and identified a significant correlation between m1A regulators and OS in 33 cancer types. Some m1A regulatory genes showed carcinogenic characteristics. For example, high expression of TRMT6 and TRMT61A was related to poor OS across different cancer types. In particular, in KICH and LIHC, ALKBH1 and TRMT6 genes showed extremely high hazard ratio (HR) values. These observations indicated that ALKBH1 and TRMT6 may have oncogenic potential. In contrast, we found that some m1A regulators also show associations with tumor suppression, such as TRMT61B. High TRMT61B expression significantly correlated with higher survival rate in rectum adenocarcinoma (READ) and kidney renal clear cell carcinoma (KIRC) (Fig. 4A). And similar results were observed in the PFI and DSS analyses (Fig. 4B, C).

Correlation between m1A regulators and OS across 33 cancer types.
Most m1A regulators were significantly associated with patient OS in KICH and LIHC. Therefore, we investigated whether m1A regulator expression could contribute to assess patient risk. Patients with LIHC and KICH were classified into two subgroups based on m1A regulator expression levels. For LIHC, the first subgroup consisted of 199 patients with high expression of m1A regulators, and the second subgroup consisted of 172 patients with low expression (Fig. 5A). For KICH, the first subgroup consisted of 28 patients with high expression of m1A regulators, and the second subgroup consisted of 37 patients with low expression (Fig. 5B). Compared with the high expression subgroups, the survival rate of low expression subgroups was significantly better in LIHC (p = 0.0041, Fig. 5C), and similar results were observed in KICH subgroups (p = 0.0034, Fig. 5D). Taken together, these results indicated that expression of m1A regulators correlate with patient OS and may serve as a prognostic marker for KICH and LIHC.

Correlation between m1A regulator expression and OS in LIHC and KICH.
m1A regulator involvement in carcinogenic pathways
To clarify the functional role behind m1A gene expression changes in tumor genesis and development, we studied the relevance between individual m1A regulatory protein expression and associated Kyoto Encyclopedia of Genes and Genomes (KEGG) pathway. We found that the expression of different m1A regulatory proteins correlated with the activation or inhibition of various carcinogenic pathways (Fig. 6A). The expression of TRMT6, TRMT10C, and YTHDF2 was related to multiple activation pathways such as the cell cycle and DNA replication and mismatch repair pathways (Fig. 6B). In particular, we observed that the m1A regulator TRMT10C was associated with a number of activated pathways, including DNA replication, ubiquitin-mediated proteolysis, and base excision repair signaling. In contrast, we found that YTHDC1 expression was related to multiple inhibitory pathways, including drug metabolism, glutathione metabolism, and neuroactive ligand receptor interaction signaling (Table 1). Moreover, m1A regulators within different functional classes could associate with the same pathways.

m1A regulators are correlated with the activation and inhibition of cancer-related pathways.
Several Carcinogenic Pathways Related to N1-Methyladenosine Regulators
Our findings also imply that writers, readers, and erasers do not function in isolation and are correlated in the context of cancer. We discovered that m1A regulator showed significantly coincident gene expression changes within and between functional classes. For example, the expression of eraser gene ALKBH1 significantly positively correlated with the writer gene TRMT10C. We also found, for instance, a high correlation for TRMT61B and YTHDC1 in the same protein complex (R = 0.51, p < 0.01) (Fig. 6C). In addition, we observed that writers, readers, and erasers can interact in the same PPI network (Fig. 6D). The interaction between readers and erasers was particularly frequent. Taken together, these results indicated that m1A regulators were associated with the activation or inhibition of multiple carcinogenic pathways, and corresponding interactions existed among writers, readers, and erasers.
Discussion
An increasing number of studies have revealed that m1A regulators play a vital role in human diseases, including cancer. For example, PCA-1/ALKBH3 contributes to pancreatic cancer by supporting apoptotic resistance and angiogenesis (Yamato et al., 2012). AlkB homolog 3-mediated tRNA demethylation promotes protein synthesis in cancer cells (Ueda et al., 2017). Liu et al. (2020a) found that YTHDF2/3 is required for somatic reprogramming through different RNA deadenylation pathways. Moreover, N-methyladenosine regulates glycolysis of cancer cells through PDK4 (Li et al., 2020) YTHDF1 as an amplifier of Wnt/b-catenin signaling at the translational level, which is required for the maintenance of intestinal stem cells (ISCs) during regeneration and tumorigenesis (Han et al., 2020). Transfer RNA demethylase ALKBH3 promotes cancer progression through induction of tRNA-derived small RNAs (Chen et al., 2019b). Li et al. (2019) also explored the landscape of m1A regulators in 33 cancers. In this study, we investigated genetic and expression alterations to m1A regulators in 33 cancer types. Uterine corpus endometrial carcinoma (UCEC) contains the most types of mutations, and the top 5 mutations were YTHDC1, YTHDF3, TRMT61B, TRMT10C, and TRMT6. Wang et al. (2020c) found out that YTHDF3 is closely associated with UCEC patient survival outcomes. In STAD, we found out that the top 3 m1A related mutations were YTHDF1, YTHDF2, and YTHDC1. In READ, YTHDC1, YTHDF3, and YTHDF1 were the top 3 m1A related mutations. In colon adenocarcinoma (COAD), we found out that YTHDC1, YTHDF3, and YTHDF2 were the most common m1A regulator related mutations. These studies indicate that m1A regulator related mutations are common in various cancers. The different mutation frequencies among pan-cancers are significantly different. Further validations are still in need to explore the m1A mutations.
In universal somatic mutations and CNV alterations were observed, which support the concept that m1A regulator dysregulation plays a key role in different cancers. Consistent with our findings, it was reported that genetic variants linked to RNA modification levels are associated with multiple disease and disease-related phenotypes, including blood pressure, hepatocellular carcinoma, breast cancer, and psoriasis (Bai et al., 2019; Han et al., 2019; Lan et al., 2019; Lin et al., 2019; Ali et al., 2020; Shi et al., 2020; Xiao et al., 2020).
The correlation between m1A regulators and OS was also evaluated, in which m1A regulators showed a significant association with patient OS across 33 cancer types. The m1A regulator related OS is remarkably different among cancers. For example, adrenocortical carcinoma (ACC), KICH, KIRC, lower grade glioma (LGG), LIHC, mesothelioma (MESO), Sarcoma (SARC), and uveal melanoma (UVM) embraced a variety of m1A regulator related mutations. In this study, we paid more attention on LIHC and KICH. Furthermore, the expression of m1A regulators might be conducive to predict risk of LIHC and KICH patients. These studies uncovered the important role of m1A regulators in cancer prognosis prediction. Similar to our research, Liu et al. (2020b) reported that most of the m6A modulators, including METTL3, METTL14, WTAP, KIAA1429, FTO, ALKBH5, YTHDF1, YTHDF2, and IGF2BP1, are upregulated and act as poor diagnostic biomarkers in patients with cancer. Zhou et al. (2019) demonstrated that deletion of m6A writer genes and copy number gain of eraser genes are independent risk factors for OS in renal clear cell carcinoma. Overexpression of YTHDF1 is associated with poor prognosis in patients with hepatocellular carcinoma (Chen et al., 2018; Zhao et al., 2018). Overall, these results indicate the promise of m1A regulators as prognosticating markers for some tumor types, which also suggests that there is potential for the development of novel clinical treatment strategies.
To further explore the molecular mechanisms of m1A regulator gene variation in cancer, we evaluated the relationship between m1A regulator expression and various cancer-related pathways. Our results showed that the expression of m1A regulators is associated with the activation or inhibition of several tumorigenic pathways. Moreover, the interactions among writers, readers, and erasers existed in cancer. Zheng et al. (2020) found that the cytoplasmic m1A reader YTHDF3 inhibits trophoblast invasion by downregulation of m1A-methylated IGF1R. Li et al. (2019) reported that the expression of m6A regulators correlates with the activation or inhibition of multiple oncogenic pathways. METTL3 may serve an oncogenic function in the progression of human ovarian cancer cells partially through the AKT signaling pathway, and it also may promote bladder cancer progression through AFF4/NF-κB/MYC signaling network (Cheng et al., 2019; Yue et al., 2019; Liang et al., 2020). In summary, our study robustly demonstrates that m1A writers, readers, and erasers may be involved in the formation of tumorigenic pathways and may be important contributors to the genesis and progression of various types of tumors.
In this study, we identified the widespread genetic and expression changes of m1A regulators in 33 cancer types. The expression of these m1A regulators contributed to the OS of tumor patients. In addition, these m1A regulators were closely related to the activation and inhibition of carcinogenic pathways. In general, this systematic analysis of the molecular characteristics and clinical significance of m1A regulators across 33 cancer types might aid in furthering our understanding of RNA methylation dysregulation and its role in cancer progression, while also providing insight into prognostication and targeted therapies. Further studies about the molecular characteristics of these m1A regulators and their mechanisms of function within specific cancer types are required.
Footnotes
Authors' Contributions
Conceptualization: J.L., and Y.C. defined the research theme. C.Z. and X.Y. drafted the article and analyzed the data. All authors read and approved the final article.
Disclosure Statement
No competing financial interests exist.
Funding Information
This study was supported by the National Natural Science Foundation of China (81702927).
Supplementary Material
Supplementary Figure S1
Supplementary Table S1
References
Supplementary Material
Please find the following supplemental material available below.
For Open Access articles published under a Creative Commons License, all supplemental material carries the same license as the article it is associated with.
For non-Open Access articles published, all supplemental material carries a non-exclusive license, and permission requests for re-use of supplemental material or any part of supplemental material shall be sent directly to the copyright owner as specified in the copyright notice associated with the article.