
Abstract
Many kinds of cancer cells are intrinsically sensitive to ferroptosis, and research interest regarding ferroptosis has been sparked by its significant role in many detrimental diseases. Ferroptosis is a novel type of iron-dependent cell death mediated by accumulation of reactive oxygen species and lipid peroxidation. Furthermore, a large number of small agents can induce ferroptosis in numerous kinds of cancer cells, including prostate cancer, pancreatic cancer, breast cancer, lymphomas, and renal cancer. These insights may help discover novel approaches for cancer therapeutic strategies; however, there is considerable uncertainty regarding ferroptosis in head and neck cancer (HNC). So far, no review of the current studies on this topic has been published. Therefore, we here elaborate the mechanisms of ferroptosis and summarize the latest findings regarding its role in HNC according to current literature. The respective findings shed light on the role of ferroptosis in HNC treatment with a number of important implications for future practice in HNC management, as outlined in this review.
Introduction
Head and neck cancer (HNC), predominantly head and neck squamous cell carcinoma (HNSCC), includes a group of tumors developing from the squamous mucosa of nasal and oral cavity, paranasal sinus, nasopharynx, oropharynx, larynx, and hypopharynx. It is the sixth most prevalent cancer type worldwide (Parkin et al., 2005). The molecular mechanisms underlying HNSCC are unclear, although some risk factors have been identified, including smoking and alcohol abuse; however, infections with oncogenic viruses also play a significant role in developing these tumors, especially human papilloma virus (Gillison et al., 2008). Annually, 600,000 new cases are diagnosed globally, with a mortality of 40–50% (Bray et al., 2018).
Clinically, an aggressive multimodality approach including surgery, radiotherapy, chemotherapy, and target therapy remains the cornerstone in the management of HNSCC. However, the 5-year survival rate of HNSCC patients has not improved significantly over the past decades, mainly because most patients are diagnosed at advanced stages of the disease and tend to suffer from a high risk of recurrence and/or distant metastasis, resulting in poor prognosis (Yanamoto et al., 2012; Lo Muzio et al., 2014). In addition, HNSCC shows intrinsic or acquired resistance to chemotherapy; therefore, it is essential to develop new strategies to enhance sensitivity of HNSCC to chemotherapeutic drugs.
At present, research on cell death mechanisms offers novel insights into human diseases. Ferroptosis, first described in 2012, is an iron-dependent programmed cell death (PCD) mechanism characterized as accumulation of reactive oxygen species (ROS) and lipid peroxidation (Dolma et al., 2003). Iron is a critical factor for ferroptosis, thus sufficient iron contributes to redox disruption as a result of system Xc− (xCT) inhibition and glutathione peroxidase 4 (GPX4) depletion (Yang et al., 2014; Angeli et al., 2017). Numerous kinds of cancers (e.g., ovarian carcinoma, adrenocortical carcinomas, diffuse large B cell lymphoma) are sensitive to ferroptosis, thus this mechanism is a promising factor regarding cancer treatments.
The main barrier for treating patients with local advanced, recurrent, or metastatic HNSCC is intrinsic or acquired resistance to chemotherapeutic drugs, especially regarding cisplatin. Of interest, a subtle link between ferroptosis and chemotherapeutic resistance was observed, and ferroptosis can reverse chemotherapeutic resistance or enhance sensitivity to chemotherapeutic drugs in HNSCC cells (Wang et al., 2008; Gorrini et al., 2013; Roh et al., 2016; Ye et al., 2019).
However, the mechanisms underlying the action of ferroptosis in reversing chemotherapeutic resistance remain poorly understood. Accordingly, understanding the molecular processes of ferroptosis and developing novel strategies for HNSCC treatment are of great significance. This review highlights some of the newest findings in this area so as to open novel perspectives for therapeutic approaches to allow better understanding and facilitate higher HNSCC treatment success rates.
Iron Metabolism and Redox Regulation
Iron, an essential mineral for facilitating cell growth and proliferation, is stored and utilized in mitochondria, and it occurs at higher concentrations in mitochondria of lung carcinoma cells (Nie et al., 2005) and rat hepatocytes (Rauen et al., 2007). Extracellular ferric iron (Fe3+) binds to the carrier protein transferrin (TF) forming diferric TF, which then binds to TF receptors (TFR) on the cell membrane. Fe3+ is dissociated from the complex by endocytosis and pH reduction. At this time, the metalloreductase six-transmembrane epithelial antigen of prostate 3 contained in the endosome converts Fe3+ to ferrous iron (Fe2+). Subsequently, Fe2+ is transported by the divalent metal transporter 1 out of the endosome and into mitochondria and is integrated in the labile iron pool (LIP), which is a transient pool of active iron (Fig. 1).
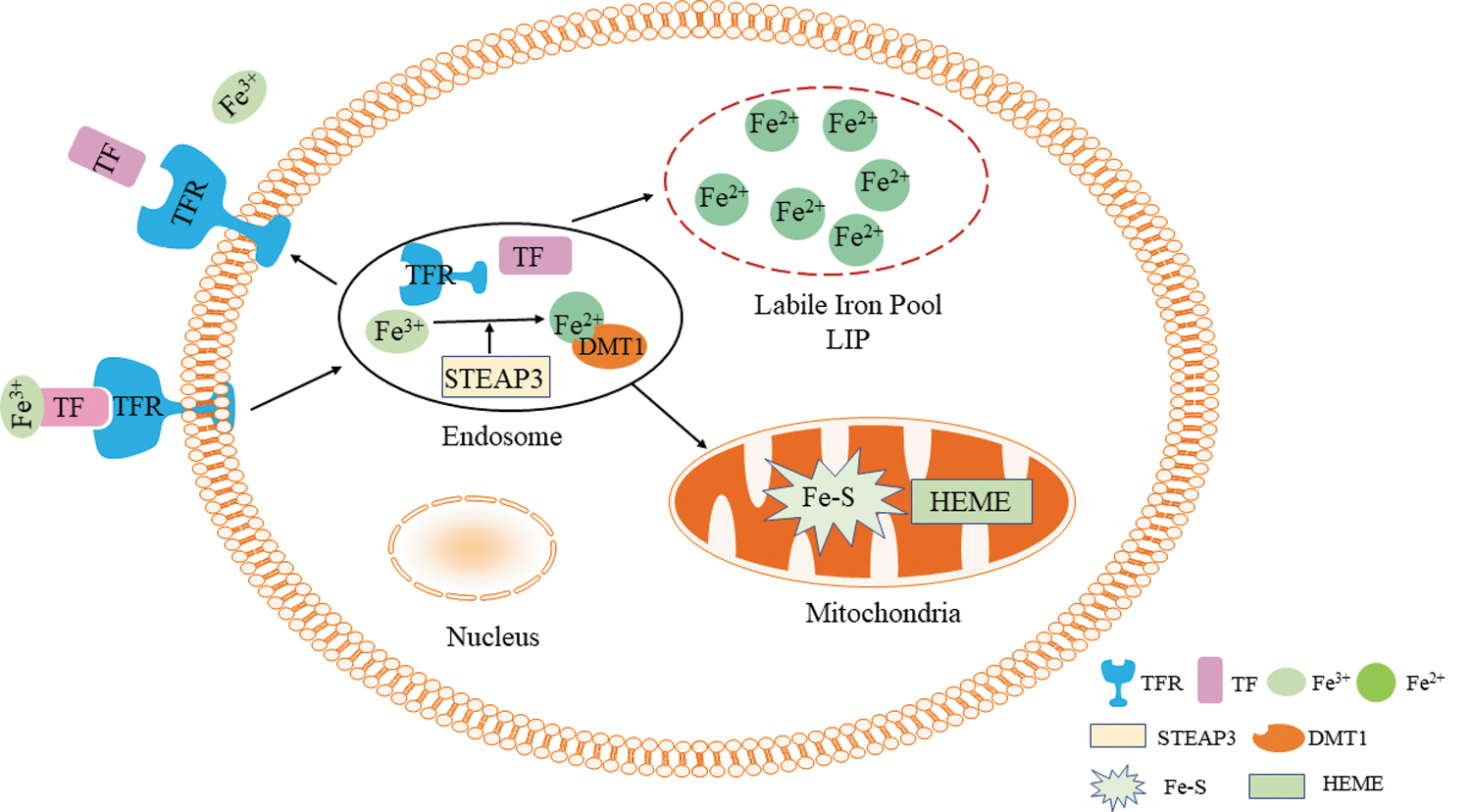
Iron metabolism. DMT1, divalent metal transporter 1; Fe2+, ferrous iron; Fe3+, ferric iron; Fe-S, iron-sulfur proteins; STEAP3, six-transmembrane epithelial antigen of prostate 3; TF, transferrin; TFR, transferrin receptors. Color images are available online.
The central role of Fe2+ is that of a cofactor in synthesis of heme, iron-sulfur (Fe-S) proteins, and a variety of other enzymes involved in redox regulation, adenosine triphosphate (ATP) generation, and other cellular processes through the Fenton reaction. Mitochondria execute a variety of functions relating to cell respiration, energy generation, biosynthesis of DNA, and other metabolic intermediates such as lipids and amino acids (Cheng and Ristow, 2013). Iron is an important player in these processes owing to its chemical properties.
Fe2+ has an unpaired electron, thus it can gain or lose one electron. As a consequence, Fe2+ shows flexible redox capability and can engage in synthesis of iron-containing proteins, which is why it is required for oxidative phosphorylation. Molecular oxygen (O2) generates “primary” ROS, superoxide anion radicals ( ) through mitochondrial respiratory chain, a reduced form of nicotinamide-adenine dinucleotide phosphate oxidase (NADPH) or xanthine oxidase. reacts with iron-containing proteins such as Fe-S proteins and heme, causing Fe2+ release and formation of “secondary” ROS (Valko et al., 2007).
) through mitochondrial respiratory chain, a reduced form of nicotinamide-adenine dinucleotide phosphate oxidase (NADPH) or xanthine oxidase. reacts with iron-containing proteins such as Fe-S proteins and heme, causing Fe2+ release and formation of “secondary” ROS (Valko et al., 2007).
Fe2+ originating from LIP and iron-containing proteins contribute to a local Fenton reaction (Fe2+ accepts an electron from iron-containing proteins, thereby converting to Fe3+). Through this reaction, hydrogen peroxide (H2O2) is reduced to OH− and lipid peroxide (LOOH) to transform lipid peroxyl radical (LOO•) or lipid alkoxyl radicals (LO•). These ROS products are crucial radicals regarding oxidative phosphorylation and cytotoxicity (Valko et al., 2007; Poprac et al., 2017; Nakamura et al., 2019), and iron is essential for these processes (Fig. 2).
Iron participates in oxidative phosphorylation and ROS generation. ┤, inhibit; →, generate; GPX4, glutathione peroxidase 4; GSH, glutathione; H2O2, hydrogen peroxide; LO•, lipid alkoxyl radicals; LOO•, lipid peroxyl radical; LOOH, lipid peroxide; NOX, nicotinamide-adenine dinucleotide phosphate oxidase; , superoxide anion radicals; ROS, reactive oxygen species; SOD, superoxide dismutase; XO, xanthine oxidase. Color images are available online.
Excessive iron can also indirectly lead to production of ROS. Iron is a crucial element in the biosynthesis of enzymes and of some iron-containing proteins that are responsible for many biological processes, for example, generation of ATP (Pratt et al., 2011; Ray et al., 2012). Overproduced ROS attack ferritin and iron-containing proteins that leads to labile iron release (Nakamura et al., 2019) and generation of further ROS.
Excessive ROS may damage macromolecular compounds, resulting in cell death through DNA degradation, lipid damage, and downregulation of protein activity; for example, •OH can react with DNA that elicits mutagenesis, carcinogenesis, and aging (Halliwell and Gutteridge, 1985). Mutagenic and carcinogenic malondialdehyde and 4-hydroxy-2-nonenal, major toxic products of lipid peroxidation, have been produced (Valko et al., 2007). Yang and Stockwell (2008) reported mechanism of cell death in 2008, which is a cause of oxidative stress owing to redox imbalances caused by ROS.
In general, cells contain numerous antioxidants to counteract effects of overly abundant ROS and to reduce ROS concentrations and to maintain a balance of ROS generation and cellular antioxidation. The produced is reduced to H2O2 by superoxide dismutase (SOD). The radical can be scavenged by GPX4 that requires an electron donated by glutathione (GSH). As cellular antioxidants, GPX and GSH can convert H2O2 to H2O and reduce the production of •OH. In addition, the antioxidant vitamin E (T-OH) is responsible for LOO•/LO• deoxidation by forming lipid hydroperoxide and T-O•, after which the vitamin E radical (T-O•) is reduced by vitamin C and GSH (Fig. 2) (Valko et al., 2007).
When concentrations of ROS (•OH and LOO•/LO• produced through the Fenton reaction) exceed the capacity of cellular antioxidation, ROS-induced oxidation of intracellular lipids, proteins, and DNA is considered the reason for accumulation of toxic biological molecules that directly or indirectly contribute to cell death (Valko et al., 2007). Therefore, dysregulation of the iron metabolism is an important elicitor of many oxidative stress-related human diseases, including malignant diseases (Bogdan et al., 2015; Manz et al., 2016), especially regarding excessive iron. In 2012, this deleterious process was termed “ferroptosis” (Dixon et al., 2012).
Mechanisms of Ferroptosis
Ferroptosis is a process of dysregulation of the iron metabolism and redox imbalance (Fig. 3). As noted previously, iron is absorbed as Fe2+ and transported in the body as Fe3+, which binds to TF and is translocated into cells by TFR, a protein located in the cell membrane. Subsequently, Fe3+ is reduced to Fe2+, which is then stored in the LIP and as ferritin (Fig. 1). Remaining Fe2+ is oxidized to Fe3+ and is discharged from the cell where it is included in further iron cycles (Manz et al., 2016). Intracellular Fe2+ and extracellular Fe3+ are typically balanced; however, when this balance is disturbed, iron intake increases, and levels of stored iron decrease. The reasons for iron overload are predominantly exogenous iron intake and endogenous iron overproduction. -mediated increase of Fe2+ by accepting an electron from Fe-S is one of the sources of endogenous iron overproduction (Fig. 2).
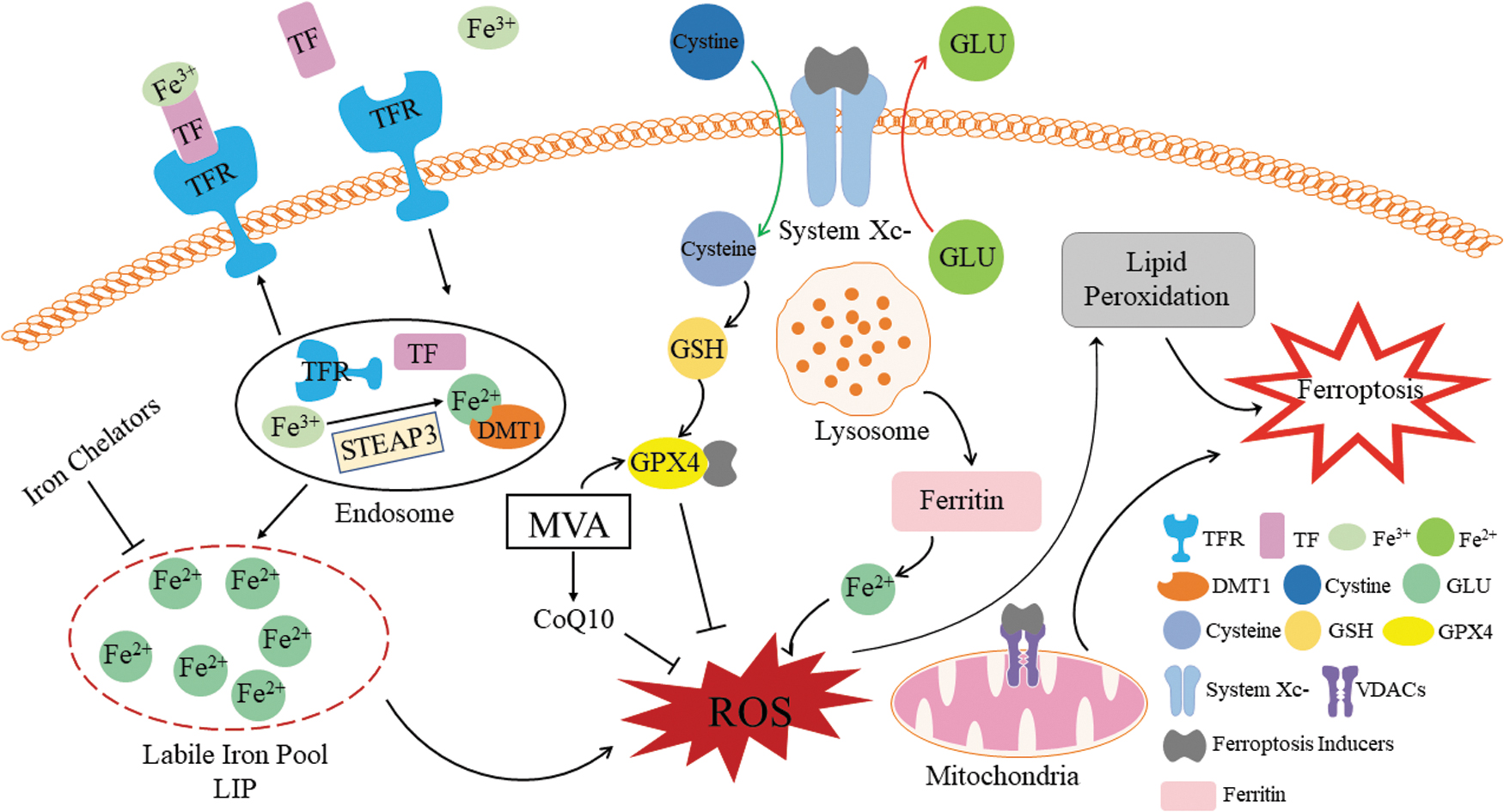
Mechanisms of ferroptosis. ┤, inhibit; →, generate; CoQ10, coenzyme Q10; GLU, glutamate; MVA, mevalonate pathway; VDACs, voltage-dependent anion channels. Color images are available online.
Apart from that, a different source is lysosomes eliciting Fe2+ increases through degradation of ferritin, which was first identified in 2014 and is frequently referred to as “ferritinophagy” (Mancias et al., 2015). Indeed, this is a part of autophagy, another phenotype of PCD; thus, some researchers suggested a correlation of autophagy and ferroptosis (Torii et al., 2016; Huang et al., 2019); this topic is further discussed hereunder.
Regarding ferroptosis, the main cause of redox imbalances is the failure of cellular antioxidative signaling pathways. In these signaling pathways, cells are equipped with many antioxidants to prevent oxidative stress caused by ROS. Of interest, each antioxidant enzyme is responsible for a specific oxide (Fig. 2).
For instance, SODs only act as superoxide scavengers. In addition to antioxidant enzymes, cells also contain numerous other nonenzymatic antioxidants, the most important of which is GSH; as shown previously, it is a cys-containing tripeptide and associates tightly with ATP-dependent reactions through the pentose phosphate pathway, and production of a reduced form of NADPH through this pathway is essential to maintain GSH in a reduced form that can protect cells from oxidative damage (Holmström and Finkel, 2014; Schmacht et al., 2017). Synthesis of GSH critically depends on activity of xCT, a membrane Na+-dependent cysteine/glutamate exchange transporter encoded by the genes SLC7A11 and SLC3A2 (Yu et al., 2017).
xCT transports intracellular glutamate out of the cell, whereas it simultaneously transports extracellular cystine into the cell, which is subsequently converted to cysteine for GSH synthesis. Oxidized and unoxidized GSH exist at a stable ratio to maintain the redox status of the cell (Schmacht et al., 2017). Evidence suggests that ferroptosis inducers markedly inhibit the capability of cysteine uptake and GSH synthesis in HT-1080 cells; however, this can be reversed by β-mercaptoethanol (Dixon et al., 2012) as it enhances cysteine uptake through different pathways (Ishii et al., 1981). In addition, when xCT is inhibited, SLC7A11 is upregulated, which is the same effect as that of ferroptosis inducers on HT-1080 cells (Lo et al., 2008). These results demonstrate the involvement of xCT in ferroptosis.
However, ferroptosis inducers can elicit ferroptosis even without exhausting GSH (Yang et al., 2014), suggesting that GSH-GPX4 is not the only pathway of ferroptosis development. GPX4 is also generated through the mevalonate (MVA) pathway. After depleting GSH, ferroptosis inducers can suppress the antioxidative ability of GPX4 and overproduce ROS, which contributes to ferroptosis (Yang et al., 2014). During this process, coenzyme Q10 (CoQ10), an effective endogenous inhibitor of ferroptosis, is produced (Shimada et al., 2016), and MVA-derived CoQ10 functions outside mitochondria to suppress lipid peroxidation by capturing radical intermediates in the process and suppresses ferroptosis.
Ferroptosis suppressor protein 1 (FSP1) is an oxidoreductase that can reduce CoQ10 and generate a lipophilic radical-trapping antioxidant that prevents lipid peroxidation and ferroptosis (Bersuker et al., 2019). This is a key component of a nonmitochondrial CoQ antioxidant system that acts in parallel to the canonical glutathione-based GPX4 pathway. Moreover, the FSP1-CoQ10 pathway of ferroptosis resistance occurs in lung cancer cells in culture and in mouse tumor xenografts, and MVA as the precursor of isoprenoid groups is used in the biosynthesis of isoprenoid end products including cholesterol that is involved in cell growth and proliferation (Goldstein and Brown, 1990; Garcia-Bermudez et al., 2019).
However, cholesterol accumulation is potentially toxic for these processes. Therefore, cells must precisely regulate biosynthesis of MVA, which is the case in the MVA pathway. Depletion of cholesterol results in apoptosis of colorectal cancer cells (Calleros et al., 2009), and squalene, an intermediate in cholesterol synthesis and in the MVA pathway, helps protect lipids from peroxidation after oxidative stress (Garcia-Bermudez et al., 2019). Lymphoma cells can benefit from accumulation of squalene, which contributes to tumor formation in vivo, whereas loss of squalene sensitizes lymphoma cells to ferroptosis inducers (Garcia-Bermudez et al., 2019). Taken together, it is reasonable to assume that regulation of ferroptosis elicited by the MVA pathway is a potential strategy for cancer treatment.
Voltage-dependent anion channels (VDACs) are transmembrane iron selective channels distributed in the outer mitochondrial membrane and are associated with nicotinamide–adenine dinucleotide hydrogen and ATP metabolism. Moreover, they are targets for some ferroptosis inducer such as erastin. For instance, erastin binds to VDACs that alters mitochondrial membrane permeability and ultimately leads to mitochondrial dysfunction and ferroptosis (Yagoda et al., 2007).
Regulatory Pathways of Ferroptosis
In addition to the mechanisms of ferroptosis summarized previously, some regulatory pathways are also engaged in regulating ferroptosis. For example, iron–sulfur clusters (ISCs) are essential for mitochondrial respiration and enzyme activity regulation. Iron–sulfur proteins are enzymes or proteins that constitute ISCs. ISCs use redox reactions with iron, thereby transforming Fe2+ to Fe3+ and vice versa (Amela et al., 2013). NEET proteins are a novel class of proteins that include nutrient-deprivation autophagy factor-1 and mitoNEET. These proteins regulate mitochondrial ferroptosis through the 2Fe-2S cluster. NEET proteins are also cellular antioxidants and negative regulatory factors of ferroptosis (Sohn et al., 2013).
Furthermore, nuclear factor-erythroid 2-related factor 2 (Nrf2) is a key transcription factor of antioxidation systems and is crucial for redox homeostasis. Nrf2 typically binds to Kelch-like ECH-associated protein-1 (Keap1) in the cytoplasm, which inhibits Nrf2 activity (Roh et al., 2017). However, when intracellular ROS concentrations increase, Nrf2 dissociates from Keap1 and enters the nucleus. Nrf2 then binds to a promoter in the nucleus and activates target genes for antioxidant response elements (AREs) (Hayes and Dinkova-Kostova, 2014). Expression of these genes reduces ROS concentrations and counteracts ferroptosis. Therefore, the Nrf2–ARE pathway is a key negative regulator of ferroptosis.
p53 is a classic tumor suppressor that protects cells from oncogenic transformation by inducing apoptosis and growth arrest in damaged cells (Levine, 1997; Vousden and Prives, 2009). Cells with p53 mutations evade cell death, and cell damage may lead to malignancy. p53 control of apoptosis involves responses to changes in mitochondrial respiration and lipid peroxidation (Maddocks and Vousden, 2011; Li et al., 2012). Thus, p53 plays a crucial role in regulating ferroptosis.
Evidence suggests that p53 promotes ferroptosis through inhibiting cystine uptake (Imai et al., 2017), thereby repressing expression of SLC7A11 (Moll and Petrenko, 2003; Jiang et al., 2015; Wang et al., 2016) and promoting SAT1 (Ou et al., 2016) and GLS2 expression (Hu et al., 2010); thus, ferroptosis may be suppressed through inactivation of dipeptidyl peptidase-4 (Xie et al., 2017) and upregulation of CDKN1A/p21 expression (Tarangelo et al., 2018). Acetylation-defective mutants of p53 (3KR, R117, R161, and R162) retain the ability to promote ferroptosis under ROS pressure (Jiang et al., 2015).
Furthermore, in addition to those classic mechanisms, cells also exhibit other antioxidant pathways. A cohort of genes antagonizing ferroptosis was identified, including GTP cyclohydrolase-1 (GCH1) and its metabolic derivatives tetrahydrobiopterin/dihydrobiopterin (BH4/BH2) through lipid remodeling (Kraft et al., 2020). The GCH1-BH4-phospholipid axis acts as a master regulator of ferroptosis resistance, controlling endogenous production of the antioxidant BH4, abundance of CoQ10, and peroxidation of unusual phospholipids with two polyunsaturated fatty acyl tails. Sufficient BH4 prevents cells from ferroptosis when GPX4 is inhibited through cystine; of interest, BH2 supplementation or GCH1 overexpression in BH4-depleted cells can lead to resistance to RSL3 treatments (Soula et al., 2020).
Moreover, recycling of quinoid dihydropteridine reductase and dihydrofolate reductase may transform BH2 to BH4, which explains previous observations (Soula et al., 2020). Taken together, this demonstrates a unique mechanism of ferroptosis protection that is independent of xCT.
Hypoxia can elicit numerous pathological processes such as tumorigenesis (Chung et al., 2010) and ischemic and inflammatory diseases (Carmeliet, 2005). During these processes, cells respond to hypoxia by regulating hypoxia-target gene expression to ensure survival.
Hypoxia-inducible factor-2 (HIF-2) is a member of the HIF family, whose responsibility is modulating the transcription of hypoxia-target genes (Dengler et al., 2014). Iron response element binding protein 1 regulates HIF-2α expression by binding to an iron response element in the HIF-2α 5′-UTR (Sanchez et al., 2007; Zimmer et al., 2008). Moreover, its stability and activity can be affected by iron chelator deferoxamine (DFO) (Befani et al., 2013). These findings shed light on the relationship of ferroptosis and HIF-2. Using CRISPR screening and lipidomic profiling, Zou et al. (2019) revealed that HIF-2α enhances susceptibility of renal clear-cell carcinomas to ferroptosis by enriching polyunsaturated lipids and activating the expression of hypoxia-inducible lipid droplet-associated protein.
In addition, the HSF1-HSPB1 pathway, MUC1-C/xCT pathways, and hemoxygenase-1 may also inhibit ferroptosis. Cells display multiple systems for maintaining cell stability and normal functions. Other pathways that remain to be identified may also be involved in the regulation of ferroptosis.
Ferroptosis and Other PCDs
Cell death occurs when a cell can no longer maintain homeostasis owing to physical, chemical, or biological effects, resulting in PCD. ROS overproduction is considered to cause cellular oxidative stress responses, and it is a well-studied cell death mechanism, as outlined previously. Given that iron has a central role in generating ROS, it may also participate in ROS production-induced PCDs such as apoptosis, necrosis, pyroptosis, autophagy, and ferroptosis. Thus, insights into similarities and differences between ferroptosis and other PCDs are helpful for better understanding ferroptosis and its relationship with cancers (Table 1).
Ferroptosis and Other Programmed Cell Deaths
GPX4, glutathione peroxidase 4; GSH, glutathione; MLKL, mixed-lineage kinase domain-like; MOMP, mitochondrial outer membrane permeabilization; RIPK1/3, regulated by receptor-interacting serine-threonine kinase 1/3; ROS, reactive oxygen species; TNFα, tumor necrosis factor α.
Apoptosis is a highly regulated process controlled by specific signaling pathways. It is characterized morphologically by nuclear and cytoplasm shrinkage, DNA degradation and condensation, and apoptotic body secretion (Taylor et al., 2008). Cytochrome c is released from mitochondria during mitochondrial outer membrane permeabilization (MOMP), a process that results in formation of a caspase-9 apoptosome and caspase-3 activation (Nakamura et al., 2019).
However, iron accumulation affects apoptosis through ROS signaling, and ROS-induced apoptosis depends on many regulatory pathways, with the predominant one occurring in mitochondria. ROS facilitates cytochrome c release through Bak/Bax during MOMP (Kuwana et al., 2002; Ott et al., 2002). Of interest, the iron-containing protein p75 NDUFS1, a subunit of respiratory complex I, amplifies the effects of ROS. Cell apoptosis is induced by increasing ROS concentrations and MOMP (Nakamura et al., 2019). In this process, the role of mediator ASK1 is crucial.
Activated ASK1 phosphorylation and MAPK pathway members activation, p38 and JNK, induce apoptosis. This ASK1–p38/JNK pathway depends on iron to be activated (Mantzaris et al., 2016). Moreover, death receptors binding their ligands is another important step of apoptosis initiation. For instance, Fas occurs in two forms in the cell, that is, a proapoptotic membrane-binding form and an antiapototic soluble form (Cheng et al., 1994), which depends on alternative splicing (Cascino et al., 1995). Excessive iron promotes alternative splicing and production of the proapoptotic form of Fas, which mainly affects human T lymphocyte populations and immune system functions (Roesler, 2005; Nakamura et al., 2019).
Necrosis is a classic PCDs and is characterized by cell swelling and membrane disruption. It is a passive cell death that is initiated by death signaling and is regulated by receptor-interacting serine–threonine kinase 1/3 (RIPK1/3) and mixed-lineage kinase domain like. Common death signaling includes TNF-α, FasL, and ROS (Berghe et al., 2014). As elaborated previously, iron regulates alternative slicing of Fas; thus, it is not surprising that necrosis can be triggered by iron through FasL when caspase-8 is inhibited (Kawahara et al., 1998) and RIPK is activated (Holler et al., 2000).
Thus, iron plays the same role in necrosis as in apoptosis. It is important to note that these two PCDs have some crossover regulatory pathways such as JNK. In addition, iron can induce necrosis directly in the form of ferritin heavy chain (FTH) and iron-containing protein heme (Xie et al., 2005; Fortes et al., 2012; Daruich et al., 2019). TNF receptor 1, Toll-like receptor 4, and heme oxygenase-1 are also required for this process (Nakamura et al., 2019).
Pyroptosis, which is elicited by inflammatory factors caspase-1/4/5/11, induces continuous enlargement of cells until rupturing, followed by recruitment of inflammatory factors to dispose cell debris. This process critically depends on the formation of plasma membrane pores by the gasdermin (GSDME) protein family members (Nakamura et al., 2019). Although pyroptosis shows some similarities with apoptosis, it has its own morphological characteristics. In contrast to apoptosis, pyroptotic cells suffer less damage and show intact nuclei (Fang et al., 2020).
Moreover, formation of pores in membranes is the main reason for enlargement of pyroptotic cells, whereas apoptotic cells show intact membranes (Brennan and Cookson, 2000; Fink and Cookson, 2006; Jorgensen and Miao, 2015). Its molecular mechanisms comprise the canonical inflammasome pathway and a noncanonical inflammasome pathway (Fang et al., 2020). They can be initiated by pathogen-associated molecular patterns and damage-associated molecular patterns, and by lipopolysaccharides, respectively. Of interest, treating melanoma with an iron agent or ROS inducer elicits cleavage of GSDME (Zhou et al., 2018), which exerts a key effect on switching from apoptosis to pyroptosis (Wang et al., 2017).
Autophagy was first proposed over 50 years ago, based on observed degradation of mitochondria and other intracellular structures within lysosomes of rat liver cells (Deter and De Duve, 1967). There are three types of autophagy, that is, macroautophagy, microautophagy, and chaperone-mediated autophagy (CMA). During macroautophagy, cytosolic components are transported by double-membrane vesicles, referred to as autophagosomes, which then fuse with lysosomes, thereby forming autolysosomes. By contrast, damaged proteins and organelles are directly engulfed by lysosome during microautophagy. During CMA, damaged proteins form a complex with chaperone proteins and are then translocated across the lysosomal membrane and can be recognized by the lysosomal membrane receptor lysosomal-associated membrane protein 2A, thereby contributing to degradation (Glick et al., 2010).
The main mechanisms of autophagy include over 30 autophagy-related proteins (Park and Chung, 2019), for example, autophagosome initial protein Beclin-1 and structural protein of the autophagosome LC3B (Liu et al., 2013). Autophagy is a complicated self-engulfing process. To respond to externally caused damage such as hypoxia or low energy concentrations, membranes of endoplasmic reticulum (ER) and/or the trans-Golgi and endosomes form phagophores using Beclin-1/VPS34.
For this process, a complex of Atg5, Atg12, and Atg16L as well as LC3 insertion are required. Autophagy will be completed by phagophore capturing of target substances, after which the autophagosome fuses with a lysosome to degrade engulfed molecules. Amino acids and other products of degradation can be transported back to the cytoplasm for reuse (Glick et al., 2010). Thus, autophagy is a recycling process. However, it remains unclear whether autophagy is a kind of cell death or a phase of cell death.
Ferritin as a cellular protein may also be targeted for autophagy. The role of the nuclear receptor coactivator 4 (NCOA4) was first discovered by Mancias et al. (2015), showing that NCOA4 participates in the binding of ferritin in autophagosomes for degradation, which is defined as ferritinophagy. NCOA4 deficiency contributes to a decrease of free iron and an accumulation of stored iron (ferritin) (Mancias et al., 2015; Bellelli et al., 2016). Intracellular concentrations of Fe2+, Fe3+, and lipid peroxide decrease, whereas levels of ferritin increased after inactivation of autophagy-related genes (Mancias et al., 2014). Therefore, ferritinophagy is a way to produce cellular labile iron for inducing cell death. In this regard, this process seems to resemble other PCDs, especially regarding ferroptosis.
Ferroptosis was initially identified when small antitumor agents were screened for oncogenic RAS mutations (Dolma et al., 2003). As mentioned previously, there are many similarities between ferroptosis and other PCDs. Hence, the relationship between them remains somewhat unclear owing to these contradictory results. It was proposed that ferroptosis can only be inhibited by iron chelation or by antioxidants, and genetic inhibition of cellular iron uptake can reverse this process (Dixon et al., 2012). This suggests that ferroptosis is an independent PCD and not a phase of other PCDs.
However, in the light of accumulating evidence, some researchers considered ferroptosis a type of autophagy-dependent cell death. Apart from ferritinophagy, lipophagy also promotes ferroptosis by increasing lipid peroxidation (Bai et al., 2018). Of importance, erastin- and RSL3-induced ferroptosis can be prevented by lysosome inhibitors in HT1080 cells (Torii et al., 2016). Moreover, the main characteristic of erastin-induced ferroptosis is increased permeabilization of lysosomal membranes, which results in lysosome death (Torii et al., 2016). CA-074ME, an inhibitor of lysosome-dependent cell death, reverses ferroptosis induced by erastin in pancreatic cancer cells (Gao et al., 2018). Based on this, we currently cannot confirm whether ferroptosis is an independent type of PCD.
Ferroptosis and HNSCC Treatments
Ferroptosis is implicated in many human degenerative diseases characterized by dysregulated cell death, including ischemia/reperfusion injury, neurodegenerative diseases, or damage induced by specific chemicals or pathogens (Zou and Schreiber, 2020). Ferroptosis during cancer has been thoroughly studied since the correlation between ferroptosis and tumor suppression was shown in studies using a p53 mutant (Wang et al., 2016). Several kinds of cancer cells exhibit intrinsic sensitivity to ferroptosis, such as ovarian carcinoma, and diffuse large B cell lymphoma. Thus, induction of ferroptosis is a promising approach to cancer treatment, especially regarding apoptosis-resistant tumors.
As the sixth most common cancer, >600,000 new HNSCC cases are reported every year (Bray et al., 2018). However, mechanisms underlying the development of HNSCC remain unclear. At present, surgery, radiation, chemotherapy, and targeted therapy in various combinations are recommended to treat HNSCC. Unfortunately, even with successful treatments, many HNSCC patients show therapy resistance, and the 5-year survival rate has improved only marginally in the past decades (Obid et al., 2019).
Ferroptosis as a tumor suppression mechanism leads to tumor growth suppression through metabolic regulation by four main pathways: dysregulation of the iron metabolism, GSH-GPX4, FSP1-CoQ10, and through the GCH1-BH4-phospholipid axis. Although the role of ferroptosis in HNSCC and its molecular mechanisms are not sufficiently clear so far, increasing evidence suggests that ferroptosis acts as a critical barrier of cancer development (Hassannia et al., 2019). These mechanisms are highly valuable for using ferroptosis in HNSCC treatments, and furthering our understanding of them is thus vital.
Mutation of p53 is a pivotal event in regulating ferroptosis during formation and development of many human cancers (Stockwell et al., 2020). The 3-KR-mutation of p53, which does not induce cell cycle arrest, senescence, and apoptosis remains capable of regulating SLC7A11 expression, inducing ferroptosis, and causing tumor growth suppression in xenograft models; in contrast, the p53 (K98) lysine mutation completely loses its ability to affect metabolic targets such as SLC7A11 and is severely defective regarding suppression of tumor growth in mouse xenograft models (Wang et al., 2016). Therefore, it appears that ferroptosis contributes to tumor suppression; however, this antitumor process may be reversed by some autophagy inhibitors (Zhou et al., 2019).
In this regard, some interesting observations were made in lymphomas cell lines: knockout of squalene synthase in ALK+ anaplastic large cell lymphoma cells results in decreased squalene levels and sensitizes it to ferroptosis inducers (Garcia-Bermudez et al., 2019). Moreover, it was confirmed that clear-cell carcinomas exhibit high sensitivity to three ferroptosis inducers (RSL3, ML120, and ML162) through the HIF pathway, particularly in ovarian clear-cell carcinoma cell lines ES-2, OVISE, and TOV21G (Zou et al., 2019). In addition, the HIF pathway is responsible for ferroptosis susceptibility in nonsmall cell lung cancer cells (Jiang et al., 2017). It is therefore necessary to elucidate the mechanisms behind ferroptosis resistance to establish barriers for tumor treatment.
The identified ferroptosis agonists are classified in two groups based on their mechanisms (Yang et al., 2014). Class I agonists can induce ferroptosis by repressing GPX4 indirectly through depletion of GSH, which is required for GPX4 synthesis. Erastin is the first identified Class I agonist. When this small molecule was used to treat tumor cells with RAS mutations, TFR1 increased while FTL and FTH1 decreased, compared with controls (Dolma et al., 2003). Thus, iron uptake increased and stored iron decreased in mutant cells. Moreover, ROS levels in HT-1080 cells are elevated and cells die after exposure to erastin (Dixon et al., 2012). Furthermore, DFO reduces and adding Fe2+/Fe3+ increases cell death, and antioxidants inhibit the induction of cell death. At present, there is no evidence of ferroptosis occurring in HNSCC cells, and erastin is not yet used in HNSCC treatments.
Owing to its antibiotic and immune suppressive activities, sulfasalazine (SAS) is an effective drug for treating Crohn's disease and rheumatoid arthritis. This compound is also a classic ferroptosis inducer (Xia et al., 2019), and it exhibits antitumor effects by inhibiting xCT, leading to depletion of GSH and decreased levels of GPX4 (Guan et al., 2009). As a class I agonist, it stimulates ferroptosis in many tumors, independent of presence of oncogenic mutations such as RAS, RAF, PIK3CA, and TP53 (Emma et al., 2014). This drug is also used for treating breast cancer with low ER expression (Yu et al., 2019). Genes of the CDGSH iron-sulfur domains (CISD) family encode Fe-S proteins responsible for cell proliferation and antioxidation (Sohn et al., 2013). Thus, these genes and their protein products are important for tumor cell resistance to ferroptosis. In HNC cells, ferroptosis can be elicited by SAS by inhibiting CISD2, a member of the CISD gene family (Kim et al., 2018).
Compounds that enhance ferroptosis by directly inactivating GPX4 are considered class II ferroptosis agonists. GPX4 is a negative regulator of ferroptosis, and its inhibitor can induce ferroptosis in tumor cells. (1S, 3R)-RSL3 is a prototypic agent. This agent is a ferroptosis inducer that is selectively lethal to tumor cells with RAS mutations (Yang and Stockwell, 2008), which is also true in other cancers, including colorectal cancer (Sui et al., 2018), embryonal cancers, alveolar rhabdomyosarcoma (Codenotti et al., 2018), and melanoma (Luo et al., 2018). Treatment with RSL3 and trigonelline promotes ferroptosis in HNC cells by inhibiting NrF2 gene expression, which increases resistance to ferroptosis (Shin et al., 2018).
The traditional Chinese medicines artesunate and dihydroartemisinin (DHA) are semi-synthetic derivatives of artemisinin that elicit ferroptosis in HNSCC cells. These drugs are first-line anti-malarial drugs that exert various biological effects including efficient and specific antitumor effects owing to induction of cell death.
Artesunate was tested on different HNC cell lines including three cisplatin-resistant lines, which showed lower efficiency in cisplatin-resistant HNC, whereas normal cells survived without damage (Roh et al., 2017). Artesunate can overcome ferroptosis resistance in HNC by inhibiting the Nrf2-ARE pathway. Furthermore, DHA arrests the cell cycle and promotes ferroptosis in HNSCC cells by binding GSH (Lin et al., 2016).
Ferroptosis and HNSCC Therapy Resistance
Many cancer cells exhibit negative responses to chemotherapeutic drugs, but this resistance can be reversed by ferroptosis. Nonsmall cell lung cancer shows resistance to cisplatin especially in wild-type EGFR cells; however, the underlying mechanisms remain unclear. Ferroptosis inducers enhance sensitivity to cisplatin in wild-type EGFR cells of nonsmall cell lung cancers by mediating ROS levels (Yamaguchi et al., 2013). Similarly, glioblastoma multiforme patients can benefit from a combination treatment of temozolomide and erastin (Chen et al., 2015). These findings suggest that ferroptosis is potentially a new strategy in cancer therapy. So far, one of challenges for HNSCC treatment is the resistance of cancer cells to chemotherapeutic drugs, which contributes to poor prognoses in most HNSCC cases.
Cisplatin is a DNA intercalator, typically used as a first-line chemotherapy for solid neoplasms. Owing to its broad antitumor spectrum and high efficacy, this compound is recommended by the World Health Organization, and it can induce multiple inter- and intrastrand crosslinks through binding with guanine residues. These crosslinks result in cell death in rapidly proliferating tissues (Modur et al., 2015). High doses of cisplatin are also used in radiosensitive tissues during radiotherapy. A meta-analysis of HNSCC chemotherapy treatments revealed that chemoradiation is associated with substantial long-term freedom from relapse (Pignon et al., 2000; Blanchard et al., 2011), 7% at 2 years and 8% at 5 years, with a hazard ratio of 0.81 (Pignon et al., 2000). However, consecutive high-dose cisplatin treatments frequently lead to resistances, which remains a significant concern for clinical practice.
Susceptibility to ferroptosis may be acquired during cell-state transitions in cancer, as is the case during development of therapy resistance (Zou and Schreiber, 2020). Increased intracellular glutathione concentration is associated with cisplatin resistance in cancer cells (Meijer et al., 1990; Godwin et al., 1992). An siRNA and shRNA used to silence the SLC7A11 gene induced glutathione depletion through inhibition of xCT in cisplatin-resistant HNSCC cells, which caused effective ferroptosis of resistant HNC cells and enhanced cisplatin cytotoxicity in resistant HNC cells (Roh et al., 2016).
Ferroptosis is currently considered an effective strategy for overcoming resistance in refractory tumors. Artesunate selectively induces ferroptosis in HNC cells while preserving normal cells owing to the result of the activation of ferrous iron and blocking xCT (Roh et al., 2017). Cancer cells frequently show Nrf2 overexpression, which is linked to increased resistance to anticancer therapies and poor survival outcomes in cancer patients (Wang et al., 2008). Artesunate sensitivity decreased in some cisplatin-resistant HNCs and ferroptosis resistance of HNCs because of Nrf2–ARE pathway activation. Thus, Nrf2 inhibitory therapy combined with artesunate may effectively kill resistant HNC cells, while has a protective effect against oxidative damage in normal tissue.
Moreover, regarding patients with metastatic hypopharyngeal squamous cell carcinoma (HPSCC), paclitaxel (PTX), which is also an effective chemotherapy drug, was commonly used in platinum pretreatment (Ley et al., 2017). However, acquired resistance owing to high dosages is a considerable barrier for its clinical application. For this reason, low concentrations of PTX are used in some cancer treatments.
Of interest, low PTX concentrations inhibit glutaminolysis-related genes that lead to tumor growth suppression in colorectal carcinoma and ovarian cancer stem cells (Shen et al., 2015; Lv et al., 2017). To the best of our knowledge, the glutamine-related pathway is crucial for ferroptosis. Low concentrations of PTX or RSL3 used separately do not elicit significant cell death, whereas combined treatments exert substantial effects on inducing ferroptosis and cell death in mp53 HPSCC (Ye et al., 2019). Moreover, low concentrations of PTX enable RSL3 to activate ferroptosis through upregulating p53 expression; thus, SLC7A11 transcription mediated by p53 may play an important role in this process (Ye et al., 2019).
Although it remains unclear how ferroptosis can reverse chemotherapy drug resistance and which molecules or pathways are key, some studies may provide indications to resolve this issue. The main reason for therapy resistance is high mesenchymal cell state in human cancer cell lines. This state is characterized as GPX4 accumulation; thus, tumor cells protect themselves from ferroptosis via a druggable lipid-peroxidase pathway (Viswanathan et al., 2017). Hence, “persister” cells are central drivers in nonmutational drug resistance, which is the main reason for failure of targeted therapies (Groenendijk and Bernards, 2014). Remarkably, such persister cells of different cancers show similar phenotypes and gene expression patterns, and they are more vulnerable to GPX4 inhibitors (RLS3 and ML210) than parental cells (Hangauer et al., 2017).
Furthermore, a decrease in GSH and NADPH and downregulation of antioxidant genes was observed in this process (Hangauer et al., 2017). This suggests that persister cells are specifically sensitive to ferroptosis; therefore, they may be a possible target for acquired drug resistance treatments. Hence, accumulation of GPX4 not only suppresses ferroptosis but also contributes to chemotherapy resistance (Ye et al., 2019). Ferroptosis inducers significantly enhance sensitivity to cisplatin in osteosarcoma cells, and STAT3 inhibitors play a similar role as ferroptosis inducers by decreasing expression of p-STAT3, Nrf2, and GPX4 (Liu and Wang, 2019).
In epithelial cells, intracellular tight connections activate the Hippo signaling pathway (Wu et al., 2019). For example, such interactions mediated by e-cadherin suppress ferroptosis by activating the intracellular Hippo signaling pathway with the Last1/2 process activated, YAP phosphorylated, and YAP removed from nucleus, whereas antagonizing this signaling axis can promote ferroptosis by upregulating target genes of YAP such as ACSL4 and TFRC, which are also ferroptosis modulators. Our research group found that in human hypopharyngeal cancer cell lines, the expression level of CEACAM7 is significantly higher in cisplatin-resistant cells than in cisplatin-sensitive cells. CEACAM7 may increase intracellular tight connections, inhibit ferroptosis by activating the Hippo signaling pathway, and lead to drug resistance of hypopharyngeal cancer. However, these preliminary results require further investigation.
Ferroptosis participation in cellular immunity is uncertain. CD8+ T cells can downregulate expression of SLC3A2 and SLC7A11 through releasing interferon gamma, impairing cellular uptake of cystine, and facilitating lipid peroxidation within tumor cells. This effect sensitizes cells to radiotherapy, and treatment with PD-L1 blockade and cystine or cysteine depletion activates antitumor activity of T cells and induces tumor cells ferroptosis in vivo (Lang et al., 2019; Zou et al., 2019). Whether this approach is applicable to HNSCC remains unclear.
Bionanotechnology has considerably improved anticancer drug delivery regarding drug availability, targeted delivery characteristics, therapeutic efficiency, and reduced side effects. Ferroptosis-driven nanotherapeutics promotes rapid development of cancer treatment, depending on the targeted delivery of ferroptosis inducers or promoters into tumor cells and promoting intracellular accumulation of ROS and lipid peroxidation in tumor cells (Shan et al., 2020). Moreover, integration of chemotherapy with ferroptosis based on nano-Drug Delivery System has opened new avenues of cancer therapy. For instance, toxic cisplatin can produce H2O2 from cascading reactions under a reduction microenvironment in tumor cell cytoplasm, and the produced H2O2 (Ma et al., 2017) may be further catalyzed by ferric ions to generate toxic hydroxyl radicals through the Fenton reaction, resulting in both apoptosis and ferroptosis of tumor cells.
Conclusions and Perspectives
Ferroptosis, a novel form of cell death, stimulates further research on tumor development and treatment. At present, few studies focus on ferroptosis during HNSCC; however, killing HNSCC cells and sensitizing chemotherapy through ferroptosis are major research focuses that will help elucidate the underlying mechanisms. Heterogeneity is common among tumor cells, including HNSCC. Some populations of cells develop alterations to prevent ferroptosis, and understanding of such alterations is crucial for screening biomarkers of sensitivity to ferroptosis. Such biomarkers will greatly expand our understanding of promoting ferroptosis in tumors, including HNSCC.
Footnotes
Authors' Contributions
Conception and design: D.Y. and X.Z. Performing the literature search: S.L. Drafting the manuscript: all authors. Approving the final version: all authors. D.Y. is responsible for the integrity of the work as a whole. All authors read and approved the final manuscript.
Acknowledgement
This research did not receive any specific grant from funding agencies in the public, commercial, or not-for-profit sectors.
Disclosure Statement
No competing financial interests exist.
Funding Information
This work was supported by the Graduate Innovation Fund of Jilin University (Grant No. 101832020CX291), the Natural Science Foundation of Jilin Province (Grant No. 20190201212JC), the Medical and Health Personnel Special Project of Jilin Province (Grant No. 2019SCZT012).