
Abstract
QKI is a vital regulator in RNA splicing and maturation, but its role in cervical cancer (CC) is little known. In this study, we found that QKI is decreased in human CC, and overexpression of QKI inhibits HeLa cell proliferation and promotes the apoptosis of cancer cells. We identified hundreds of endogenous QKI-regulated alternative splicing events (ASEs) and differentially expressed genes (DEGs) in QKI-overexpressed HeLa cells by RNA-seq and selectively validated their expression by quantitative reverse-transcription polymerase chain reaction. The gene ontology and Kyoto encyclopedia of genes and genomes (KEGG) enrichment analysis showed that QKI-regulated ASEs and DEGs were closely related to cancer, apoptosis, and transcriptional regulatory functions. In short, QKI may affect the occurrence and development of CC by regulating gene expression through AS.
Introduction
Cervical cancer (CC), the second most common female malignancy in terms of both morbidity and mortality, poses a serious risk to women's health worldwide (Ginsburg et al., 2017). More than 569,847 new cases with CC are recorded worldwide every year, resulting in 311,365 deaths (Bray et al., 2018). At present, traditional methods such as surgery, radiation, and chemotherapeutic therapy are mostly applied in clinical treatment (Li et al., 2016b). However, a significant difference has been observed in clinical outcomes among different patients, which is difficult to predict (Gupta et al., 2018). Therefore, in-depth exploration of the molecular mechanism of CC and the search for more effective and specific therapeutic methods and targeted therapeutic drugs are particularly important for improving the treatment effects and quality of life of patients.
In a large part of human genes, the transcribed pre-mRNAs are processed into different splicing isoforms by utilizing the different splice sites, known as alternative splicing (AS) (Ariel and Crespi, 2017). AS is a key mechanism to adjust gene expression and proteome diversity and is also a critical reason for the vast difference in the eukaryotic gene and protein quantity (Blencowe, 2017; Liu et al., 2017). This form of post-transcriptional regulation is widespread in eukaryotic cells. It is estimated that more than 90% of genes in human beings will produce different mRNA isoforms through AS, which greatly increases the economic practicability of genetic information (Baralle and Giudice, 2017). The common forms of AS in human tissues contain intron retention (IR), alternative 5′ splice site (A5SS), alternative 3′ splice site (A3SS), exon skipping (ES), cassette exon (CE), mutually exclusive exon (MXE), etc. (Sultan et al., 2008). AS affects cell development, including sex determination (Planells et al., 2019), axonal guidance (Paronetto et al., 2016), cell activation, and contraction (Nakka et al., 2018). Aberrant AS is also closely allied with proliferation, apoptosis, migration, invasion, and drug resistance of tumor cells. It is reported that some splicing products are specifically expressed in cancerous tissues, which can serve as biomarkers for the disease and targets for disease treatment (Martinez-Montiel et al., 2018).
RNA-binding protein Quaking (QKI) pertains to a highly conserved signal transduction and activation of the RNA (STAR) family. QKI regulates AS by selectively interacting with the QKI response element (QRE; ACUAAY[N1–20]UAAY) located in the 3′ untranslated region (3′UTR) (Galarneau and Richard, 2005) of target pre-mRNAs. As far as we know, many cancer-related genes have been regarded as the targets of QKI, including CTNNB1 (Ji et al., 2013), FOS (Yang et al., 2011), KIP1 (Yamagishi et al., 2016), and TP53 (Subasic et al., 2016). Recently, many researchers have found that the low expression of QKI is associated with the occurrence and development of various human cancers, such as glioblastoma (Shingu et al., 2017), prostate (Wu et al., 2019), colon (He et al., 2015), lung (de Miguel et al., 2016), gastric cancer (Li et al., 2016a), bladder cancer (Shi et al., 2019), and clear cell renal cell carcinoma (Shi et al., 2020). This evidence indicates that QKI may serve as a tumor suppressor in various human cancers. Moreover, significant downregulation of QKI expression has also been reported in CC. QKI was involved in regulation of miR-574-5p-induced CC cell progression and metastasis (Tong et al., 2020). Li et al. (2017) reported that Dichloroacetate was relatively ineffective in CC cells, unlike the observations in other cancer types, due to increasing COX2 mRNA stability by downregulating QKI. However, the specific mechanism of QKI in CC has not been fully revealed.
In this study, we evaluate the QKI expression of 306 CC tissue samples derived from The Cancer Genome Atlas (TCGA). Subsequently, for the median QKI expression, we classified 306 cancer samples into two groups: high and low QKI expression and identified QKI-regulated ASEs. In addition, we discussed the potential effect of QKI in regulating AS in HeLa cells utilizing the pIRES-hrGFP-1a vector to construct the QKI overexpression model. The RNA-seq and bioinformatics analysis results confirmed that QKI could inhibit cell proliferation and promote apoptosis of HeLa cells by regulating the expression and AS of pre-mRNAs from several hundred genes involved in transcription control, cancer, and cell apoptosis signals. Overall, our research suggested that QKI regulates the AS of many important genes related to tumorigenesis and development, extending the functional importance of QKI in regulating gene expression and protein diversity at the AS level in response to various stimuli.
Materials and Methods
Cloning and plasmid construction
Primer sequences used for the Hot Fusion were determined with CE Design V1.04. Each primer pair comprises a fragment of gene-specific sequence and a 17–30 bp sequence of the pIRES-hrGFP-1a vector.
F-primer: agcccgggcggatccgaattcATGGTCGGGGAAATGGAAAC
R-primer: gtcatccttgtagtcctcgagGTTGCCGGTGGCGGCTCG
The pIRES-hrGFP-1a vector was digested with BamHI and XhoI (NEB) at 37°C for 2–3 h. Then, restriction enzyme-digested vector was run on 1.0% agarose gel and purified by Qiagen Column Kit. Total RNA of cells was extracted by TRIzol® Reagent Kit (Ambion). Reverse transcription of purified RNA and polymerase chain amplification of target gene cDNA were achieved by reverse-transcription polymerase chain reaction (RT-PCR). The recombinant plasmid pIRES-hrGFP-1a/QKI was constructed using ClonExpress® II One Step Cloning Kit (Vazyme) to carry out ligation reaction between enzyme-digested vector and cDNA of the target gene. Then, the transformation of plasmid DNA into Escherichia coli strain (DH5a) was processed by chemical preparations. E. coli transformed by recombinant plasmid was grown in Luria Bertani (LB) agar plates (1% tryptone, 0.5% yeast extract, and 1% NaCl) with 100 μg/mL ampicillin and incubated overnight at 37°C. Transformants were picked for colony PCR (28 cycles) with universal primers after blue-white colony selection. The accuracy of cloning was validated by Sanger sequencing.
Cell line and treatments
The human cervical cancer (HeLa) cell line was purchased from the Chinese Academy of Sciences Cell Bank (the STR certification is in Supplementary File S1). Cells were cultured in DMEM with 10% fetal bovine serum and 1% penicillin and streptomycin (Sigma-Aldrich, St. Louis, MO). This study is conducted in vitro without contact with patients or animals. Therefore, IRB approval is not required.
Cells were starved in a serum-free medium for 2 days. Once synchronized, the cells were seeded into multiwell plates. The cells were transfected with pIRES-hrGFP-1a/QKI recombinant vector or an empty carrier using Lipofectamine 3000 (Invitrogen) when reaching 70% cell confluence. When the transfection efficiency is more than 80%, the next step is carried out.
Assessment of gene overexpression
Glyceraldehyde-3-phosphate dehydrogenase (GAPDH) was used as a reference gene for assessing the overexpression of QKI (Cheung et al., 2014; de Campos et al., 2018). Total RNA was isolated from cells with TRIzol® Reagent Kit (Accurate Biology, Hunan, China,
The total protein extract of cells was loaded on 12% sodium dodecyl sulfate–polyacrylamide gel electrophoresis and then blotted onto a nitrocellulose membrane with a wet blotting system. After blocking, incubation was performed with primary antibodies in blocking buffer (1:1000 for QKI; Abcam, Cambridge, MA,
MTT assay
The MTT Assay Kit (Abcam) was used following the manufacturer's instructions to evaluate cell proliferation. Cells were seeded in 96-well plates at a concentration of 104 cells per well and were cultured at 37°C for 48 h after transfection. In addition, 20 μL MTT solution (5 mg/mL with PBS) was added to each well and incubated for another 4 h. Then, the supernatant was discarded, and 150 μL dimethyl sulphoxide (DMSO) was added to each well and placed on a shaker for 10 min to fully dissolve the colored formazan crystals generated from MTT. Finally, the OD values were read at 490 nm.
Flow cytometric analysis of apoptosis
Briefly, the cells (5 × 104) were inoculated into 24-well plates. After transfection, the cells were incubated at 37°C for 48 h, and viable cells were harvested and washed twice with PBS. Viable cells were double stained with fluorescein isothiocyanate (FITC)-conjugated Annexin V and 7-amino actinomycin D (7-AAD) (4A Biotech Co., Ltd. Beijing, China). Finally, cells were analyzed by applying a FACSCalibur system after the addition of 400 μL binding buffer (a buffer solution to increase the nucleic acid binding activity of the washing column).
RNA extraction and high-throughput sequencing
Total RNA was extracted from HeLa cells with TRIzol and purified by phenol–chloroform and isopropanol. The quantity and quality of the purified RNA were evaluated using the OD value at 260 and 280 nm measured by SmartSpec Plus (BioRad). RNA-seq can be carried out when the sample content ≥5 μg, the sample concentration ≥200 ng/μL, and the sample purity: OD260/280 = 1.8–2.2.
For every sample, 1 μg of the total RNA was used to prepare the RNA-seq library by VAHTS Stranded mRNA-seq Library Prep Kit (Vazyme). Polyadenylated mRNAs were purified and fragmented, as well as converted, into a double-strand cDNA. After the process of end repair and A tailing, the DNAs were ligated to VAHTS RNA Adapters (Vazyme). Purified ligation products corresponding to 200–500 bps were digested by heat-labile UDG, and the single-strand cDNA was amplified, purified, quantified, and kept at −80°C before sequencing. Finally, the libraries were operated according to the manufacturer's instructions and applied to the Illumina HiSeq X Ten system for 150 nt paired-end sequencing.
RNA-Seq raw data cleaning and alignment
RNA-seq raw data were processed using the FASTX-Toolkit (Version 0.0.14,
Differentially expressed gene analysis
The R Bioconductor package edgeR (Robinson et al., 2010) was used to screen the differentially expressed genes (DEGs). The level of gene expression was evaluated by FPKM. A false discovery rate (FDR) <0.01 and fold change ≥2 or ≤0.5 were regarded as the cutoff criteria to identify DEGs.
Alternative splicing analysis
The alternative splicing events (ASEs) and regulated alternative splicing events (RASEs) in the samples were defined and quantified through the ABLas pipeline (
Subsequently, based on the model reads of samples and alternative reads, Fisher's exact test was used to obtain the significant p-value. We determined the altered ratio of alternatively and constitutively spliced reads between contrapositive samples as the RASE ratio. The p-value < 0.05 and RASE ratio > 0.2 were regarded as the criteria for RASE detection. For repeated comparison, Student's t-test was performed to evaluate the significance of the RASE ratio. Those events which were significant at a p-value of 0.05 were considered RASEs.
Functional enrichment analysis
To pick out functional categories of DEGs, a stand-alone software DAVID and a web server KOBAS 2.0 were applied to identify gene ontology (GO) terms and enriched Kyoto encyclopedia of genes and genomes (KEGG) pathway (Xie et al., 2011). The Hypergeometric test and Benjamini–Hochberg FDR controlling procedure were used to evaluate each pathway's degree of enrichment (corrected p-value <0.05).
Reverse transcription qPCR validation of DEGs and RASEs
In this research, to verify the reliability of the RNA-seq results, a qRT-PCR was conducted for some of the DEGs identified. The information of primers is presented in Supplementary File S2. The methods of extraction and purification of total RNA from HeLa cells, reverse transcription, and amplification are the same as those described previously. Each sample needed to be repeated thrice for PCR amplification. The RNA expression levels of all the genes were calculated with the 2−ΔΔCT method, and GAPDH was used as the reference gene.
Meanwhile, the qRT-PCR assay was also conducted to validate the effectiveness of ASEs in HeLa cells. The primers for detecting ASEs are shown in Supplementary File S2. To detect alternative isoforms, we used a boundary-spanning primer for the sequence encompassing the junction of constitutive exon and alternative exon, as well as an opposing primer in a constitutive exon. The boundary-spanning primer of the alternative exon was determined based on “model exon” to detect model splicing or “altered exon” to detect AS.
Downloading RNA-seq raw data of CC from TCGA
The gene expression profile and clinical data of CC were downloaded from the TCGA database (
Results
Expression of QKI is downregulated in CC as revealed by the TCGA database
To understand the basics of the role of QKI in CC, we first analyzed the QKI gene expression level between CC specimens (306 cases) and normal controls (3 cases) in a publicly available CC dataset (TCGA). As shown in Figure 1A, the expression of QKI mRNA in CC tissue samples was significantly decreased compared with the 3 normal tissue samples. In addition, low expression of QKI in CC was also confirmed by supplementary analysis of 13 normal samples from the GTEx database (
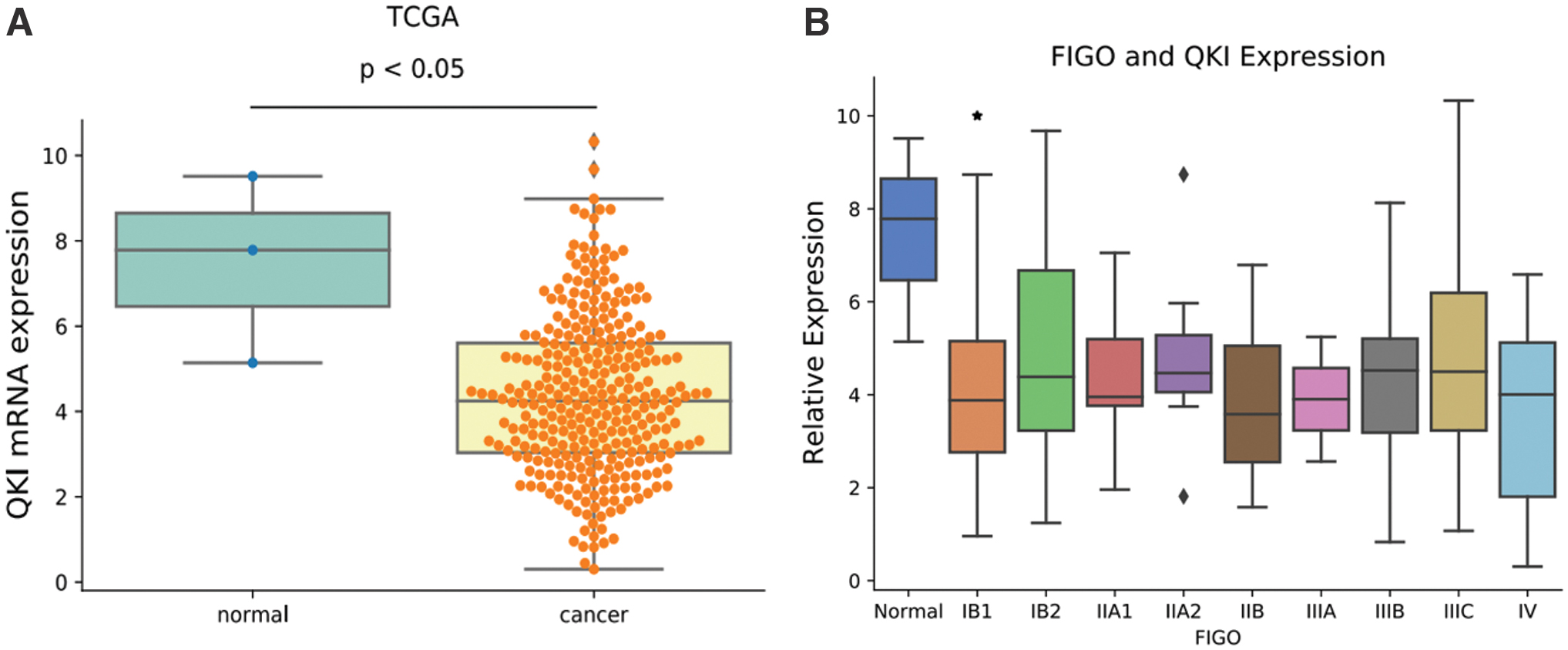
Decreased QKI expression is associated with occurrence and progression in human CC;
Potential QKI-RASEs and gene function of CC clinical samples
To clarify the possible ASEs regulated by QKI in clinical CC tissues and the life functions undertaken by involved genes, we divided all CC patients into two groups based on the median of QKI expression: low- and high-QKI group (Fig. 2A). We selected 30 patients from each of the two groups. The ABLas software was used to analyze the alteration of ASEs between patients with high- and low-QKI expression, thus finding potential QKI-regulated ASEs. Then, GO function analysis and KEGG pathway annotation were performed on the genes involved in the ASEs regulated by QKI.
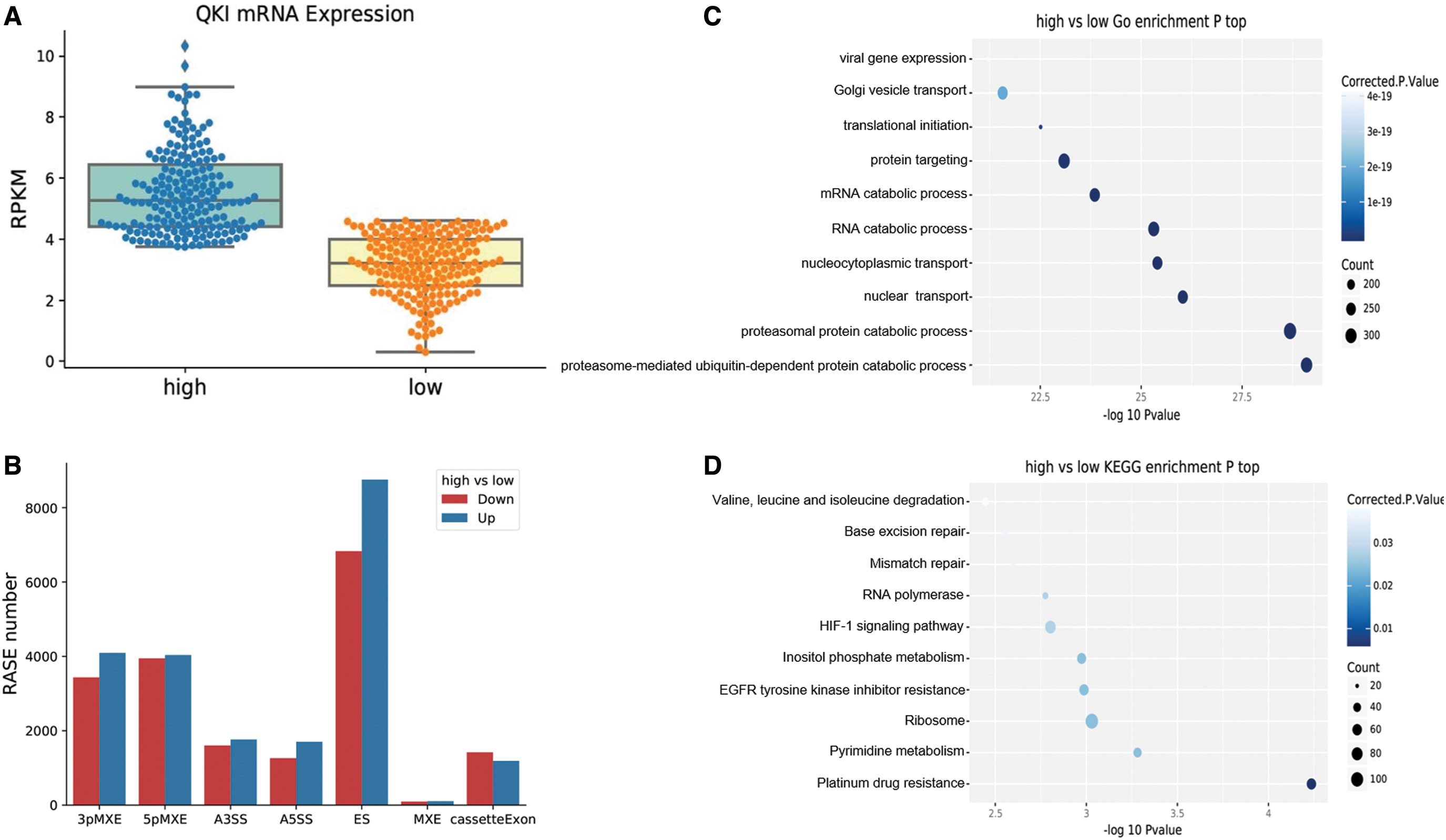
Potential QKI-regulated ASEs in clinical samples of cervical cancer.
By setting a strict criterion of p-value < 0.05, RASE ratio > 0.2, we found 40,521 QKI-regulated ASEs in two groups of CC patients (60 cases), of which 21,654 were upregulated and 18,597 were downregulated, without counting the events of IR. It could be seen that QKI-regulated ASEs were mainly distributed on ES (up 8756, down 6828), mutually exclusive 3′ UTR (3pMXE, up 4095, down 3436), and mutually exclusive 5′ exons (5pMXE, up 4038, down 3948) (Fig. 2B). These data suggest that the RNA-binding protein QKI widely regulated ASEs in CC. The genes involved in QKI-regulated ASEs were highly enriched in the GO biological process in terms of protein and RNA catabolic process, nuclear and nucleocytoplasmic transport, protein targeting, translational initiation, Golgi vesicle transport, and viral gene expression (Fig. 2C). Enriched KEGG pathways included those referred to in platinum drug resistance, pyrimidine metabolism, EGFR signaling pathway, HIF-1 signaling pathway, inositol phosphate pathway, RNA polymerase, and mismatch repair (Fig. 2D). These results indicate that QKI could regulate gene expression and substance transport in cancer-related pathways through AS, thus affecting the epigenetics of tumor tissues (All the details about the analysis of CC clinical samples can be seen in Supplementary File S3).
Upregulation of QKI expression in HeLa cell reduces cell proliferation and promotes apoptosis
Besides analyzing the above clinical data of CC downloaded from the TCGA database, we also established a functional cell model to explore the role of QKI in CC. In the early stage of the experiment, we observed low expression of QKI in CC cells by comparing the QKI mRNA levels of 28 endometrium cell lines and 2 CC cell lines downloaded from the CCLE website (

Upregulation of QKI in HeLa cells and its effects on cell proliferation and apoptosis.
QKI-regulated transcriptional difference by RNA-seq analysis
To study transcriptional regulation mediated by QKI, we performed an RNA-seq experiment. We constructed four cDNA libraries made of the abovementioned control and QKI overexpression cells (two biological replicates in each group), which were sequenced using the HiSeq X Ten system for 150 nucleotide paired-end reads per sample. We got an average of 88.0 ± 3.5 M high-quality reads of each sample after removing adaptors and contaminated sequences (details can be seen in Supplementary File S5). Then, 79.9 ± 2.4 M paired-end reads of each sample were mapped to the human GRCh38 genome, and ∼96.26–97.03% of them were uniquely mapped. To compare the gene expression patterns of these four samples, we reevaluated quantified genes and transcripts using Cuff links (Trapnell et al., 2010). We estimated the expression value based on fragments per kilobase of transcript per million mapped reads (FPKM) and got the expression levels of 28971 genes from RNA-seq (details can be seen in Supplementary File S6). Among them, the RNA-seq results further validated the amplification of QKI (Fig. 4A). FPKM values of a total of 28971 genes were subjected to a correlation matrix according to Pearson's correlation coefficient. The diagonal of the heat map represented the Pearson correlation between QKI-OE and control cells, in which the correlation matrix was symmetrical, and the two biological replications were closely correlated (Fig. 4B).
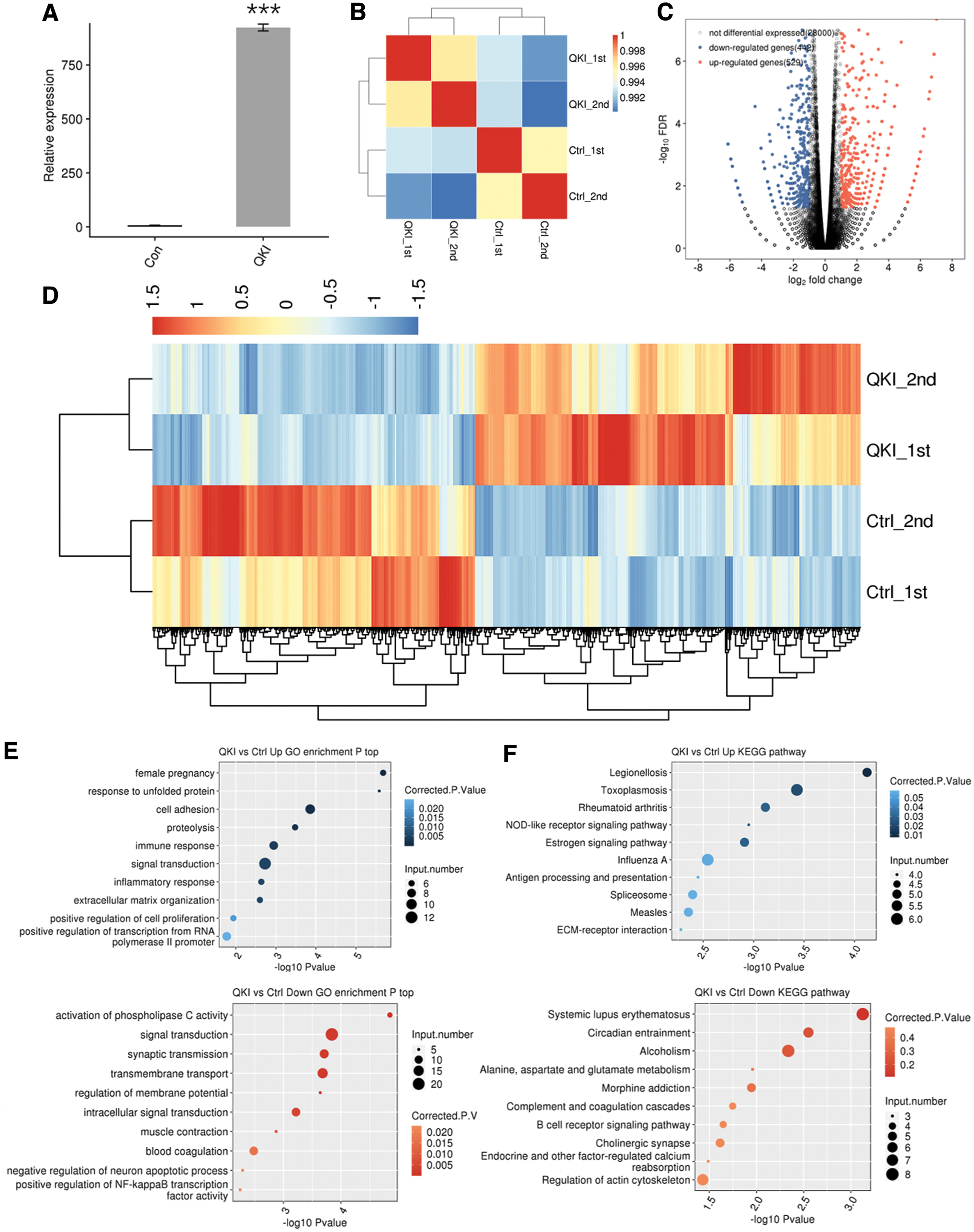
RNA-seq analysis of QKI-regulated transcriptome.
Subsequently, we applied the edgeR tool to analyze DEGs between the QKI-OE and control HeLa cells to find out genes that QKI might regulate at the transcriptional level. When the cutoff value was set as fold change ≥2 or ≤0.5 and p-value < 0.01, we found 971 DEGs, of which 529 genes were upregulated and 442 downregulated (Fig. 4C and Supplementary File S7). The clustering pattern of DEGs in RNA-seq samples showed that the transcription mediated by QKI in both data sets was highly consistent (Fig. 4D).
To reveal the function of these 971 DEGs, GO term enrichment analysis was conducted with the online software DAVID. Based on the cutoff criteria, up- and downregulated genes were enriched in 45 and 92 GO terms, respectively. In the aspect of biological process term, the 529 upregulated genes were mostly bound up with female pregnancy, cell adhesion, proteolysis, immune response, and signal transduction (Fig. 4E, upper). The 442 downregulated genes were mostly enriched in signal transduction, synaptic transmission, and transmembrane transport (Fig. 4E, lower). KEGG pathway analysis was then used to identify pathways for these 971 DEGs. A total of 115 and 159 significantly enriched pathways for up- and downregulated genes, respectively, were identified. Upregulated genes are mainly enriched in Rheumatoid arthritis, estrogen signaling pathway, and inflammatory signaling pathways triggered by some pathogens (Legionellosis, Toxoplasmosis, and influenza A virus) (Fig. 4F, upper). Downregulated genes are enriched in systemic lupus erythematosus, circadian entrainment, alcoholism, morphine addiction, complement, and coagulation cascades (Fig. 4F, lower) (details can be seen in Supplementary File S8). The results indicated that QKI could regulate the expression of a great number of genes at the transcription level. These genes are mainly involved in substance transport and signal transduction, which is closely related to autoimmune, inflammatory, and gynecological diseases.
Validation of identified QKI-regulated DEGs using qRT-PCR
Subsequently, we selectively verified several DEGs (AVPR2, C5AR1, and TP73) with significant differences identified in the RNA-seq through qRT-PCR. AVPR2 gene encodes arginine vasopressin receptor 2, which is mostly located in the distal convoluted tubule and collecting ducts of the kidney and is stimulated and activated by arginine vasopressin released from the pituitary gland to promote urine concentration and maintain water and salt balance in the organism. Some studies suggested that AVPR2 is also expressed in fetal lung tissue and may play a role in alternative splicing-related lung cancer (Wang et al., 2019a). C5AR1 gene encodes a 7-transmembrane domain G protein-coupled receptor, whose primary property is to respond to the corresponding ligand, complement factor C5a. The interaction between the C5a receptor (C5aR) and C5a may be linked to innate immune response and apoptosis (Ward, 2008; Pandey et al., 2020). Tumor protein p73 (TP73) belongs to the p53 family of transcription factors, which has functions in angiogenesis during cancer, germ cell maturation, and neurodevelopment and ciliogenesis (Nemajerova and Moll, 2019). As shown in Figure 5, the qRT-PCR results showed that the expression of AVPR2, C5AR1, and TP73 in QKI-OE HeLa cells was downregulated compared with negative control cells, which complied with the results of RNA-seq. The detailed statistical analysis is shown in Supplementary File S9. These research findings combined with our data analysis indicated that QKI regulates the expression of genes related to cancer and apoptosis.
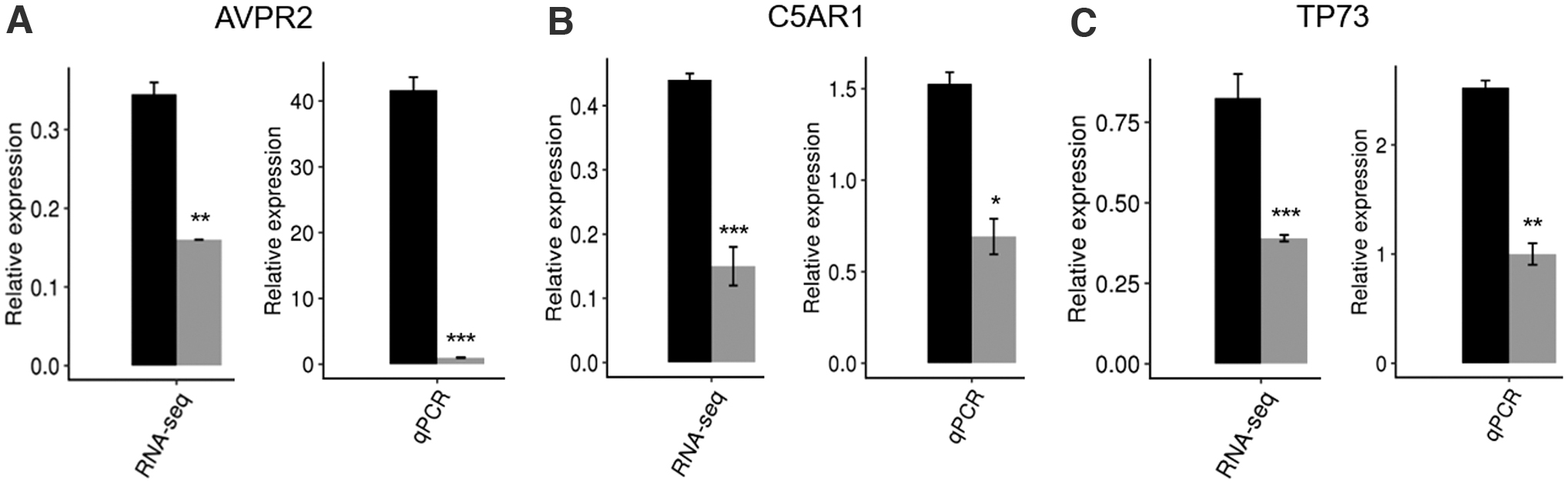
Validation of QKI-regulated DEGs in HeLa cell. The mRNA levels of AVPR2
QKI-regulated alternative splicing profiles
To uncover the role of QKI in regulating AS, we continued to analyze RNA-seq reads to investigate the QKI-dependent ASEs in HeLa cells. A total of 77.2 ± 2.3 M uniquely mapped reads were gained from QKI-OE and control HeLa cells, of which about 41.14–42.26% were junction reads (Supplementary File S5). We detected 68.9% of annotated exons (253,069 out of 367,321 annotated exons), compared these uniquely mapped reads with the referenced genome annotation, and detected 167,265 annotated splice junctions and 252,266 novel splice junctions using TopHat2. Then, we utilized ABLas software to analyze ASEs and investigate the alteration of AS occurrence. We obtained 21,964 annotated ASEs and 63,281 novel ASEs (Supplementary File S10).
By setting up strict criteria of p-value < 0.05 and RASE ratio > 0.2, we discovered 971 high-confidence RASEs involving 832 genes (Supplementary File S11). RASEs detected mainly included alternative 5′ splice sites (A5SS, 165 events), exon skipping (ES, 160 events), cassette exon (CE, 159 events), and alternative 3′ splice site (A3SS, 143 events) (Fig. 6A). These data showed that QKI widely regulates ASEs in HeLa cells and contributes to the upregulation of multiple types of ASEs more than downregulation. In addition, we found through an overlap that there were 15 genes (11 protein-coding genes, 3 antisenses, and a lincRNA) whose AS and gene expression were simultaneously regulated by QKI, suggesting that the AS and gene expression regulation of QKI in HeLa cells may be coordinated (Fig. 6B and Supplementary File S12).
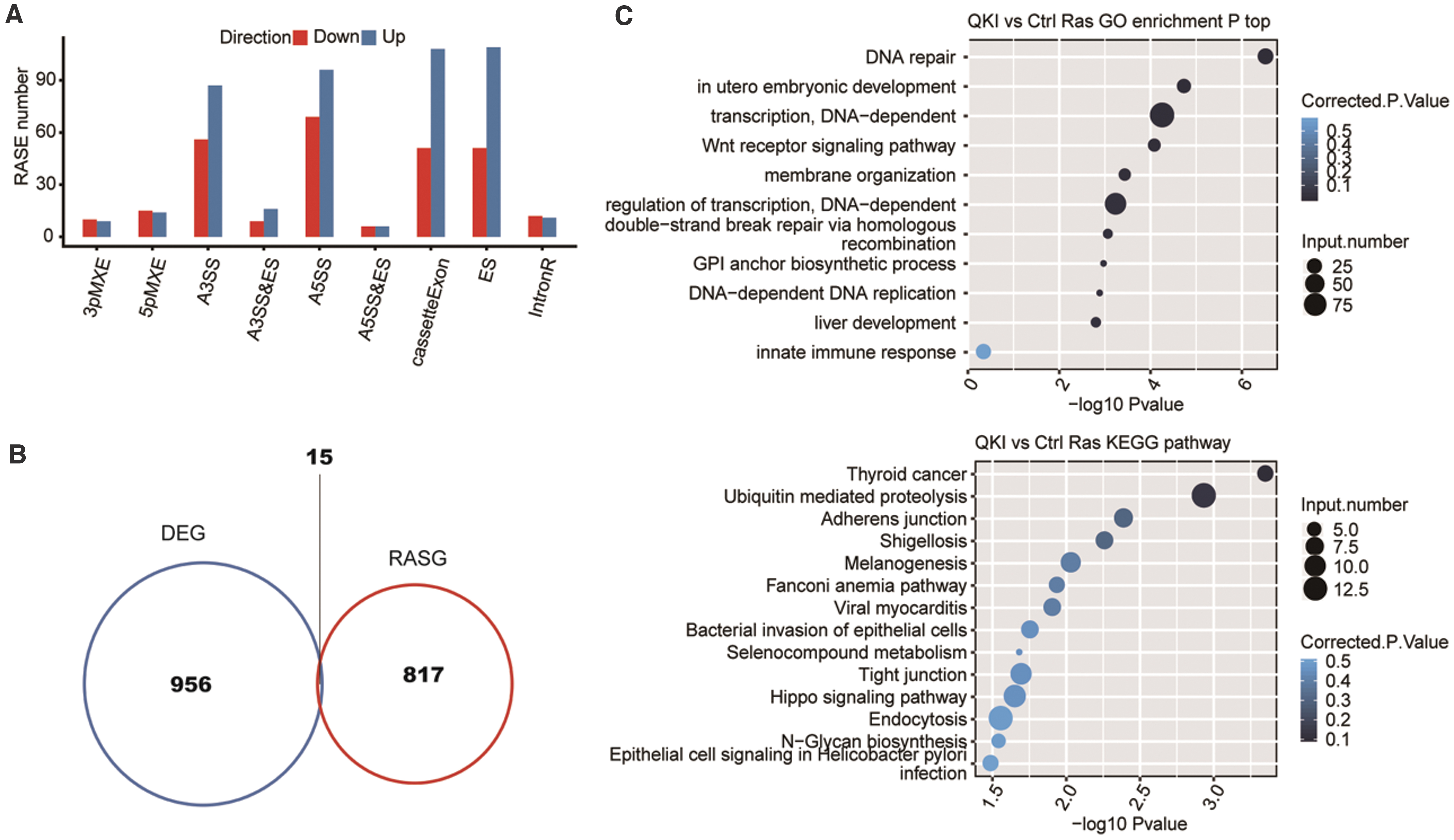
QKI-regulated alternative splicing profiles.
We further observed that those ASEs regulated by QKI were closely correlated with DNA repair, embryonic development, transcription regulation, Wnt receptor signaling pathway, and DNA replication according to GO analysis of the biological process term (Fig. 6C, upper). The most significantly enriched KEGG pathways contained thyroid cancer, ubiquitin-mediated proteolysis, melanogenesis, and the Hippo signaling pathway (Fig. 6C, lower and Supplementary File S13). These results can well echo the function of QKI-regulated ASEs in cancer.
Validation of QKI-regulated ASEs using qRT-PCR
To further verify the role of QKI-regulated ASEs in cancer, we then used another method, qRT-PCR, to selectively validate the ASEs located in 14 genes closely related to cancer. Most of these selected genes are directly or indirectly involved in the occurrence and development of tumors by encoding kinases, transcriptional regulators, or adaptor proteins (Kalogeropoulou et al., 2010; Ikeda et al., 2019; Liu et al., 2020; Wang et al., 2020a, 2020b; He et al., 2021). Design details of PCR primer pair can be seen in Supplementary File S2. Out of the 14 tested ASEs, after eliminating the unavailable data due to too many cycles, 6 genes whose AS trends were validated using qRT-PCR were consistent with RNA-seq results and had significant statistical differences: HRAS, TCF7L2, CTNND1, TAF4B, MUC16, APEX1. There were four genes whose AS trends were consistent with RNA-seq results but with no significant difference: RELA, TPM3, KRAS, SMAD2 (Fig. 7 and Supplementary File S14).
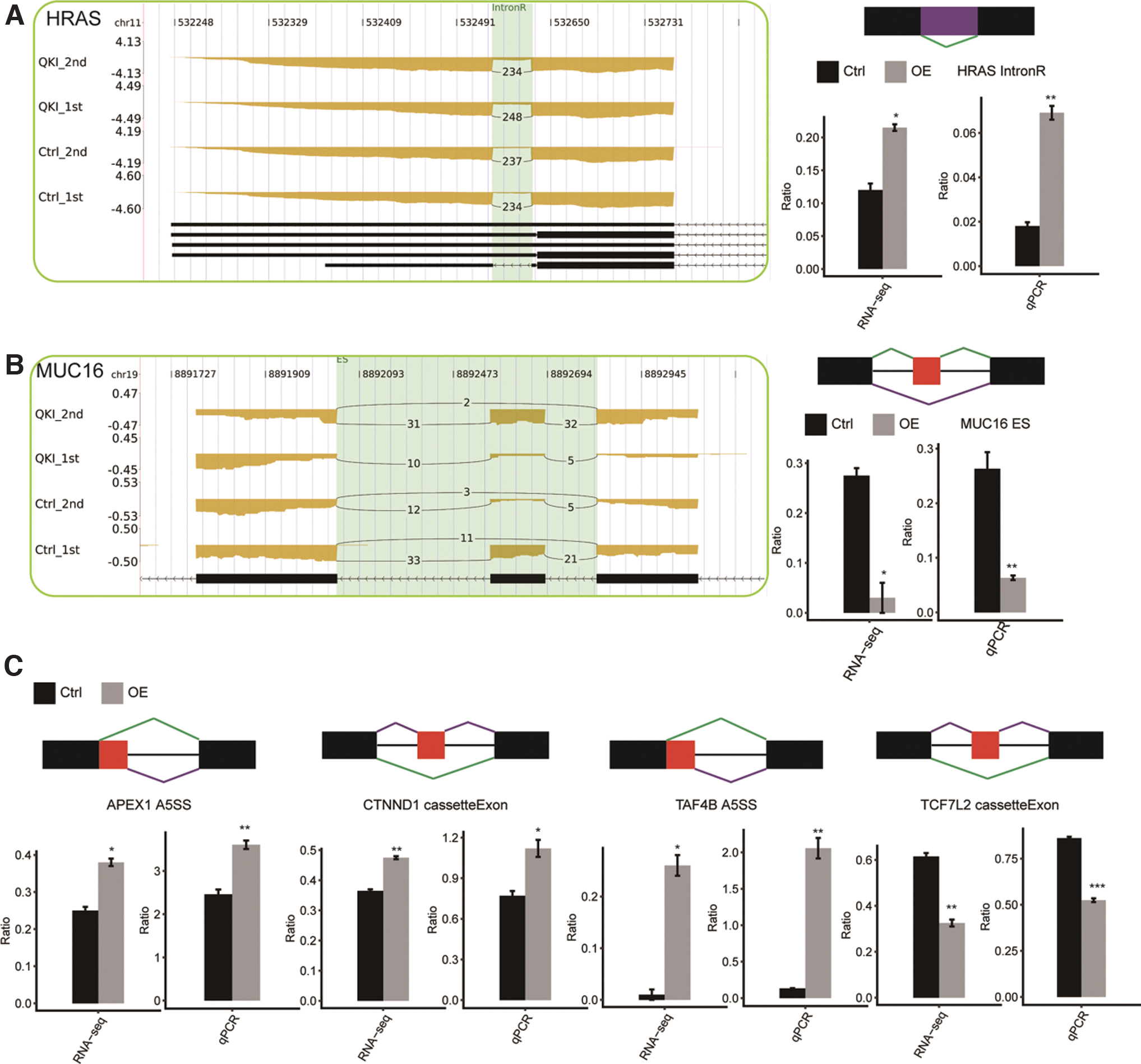
Validation of QKI-regulated alternative splicing events in cancer-related genes.
Discussion
The Quaking gene encodes an RNA-binding protein QKI containing the RNA-binding domain KH. QKI belongs to the STAR protein family with signal transduction specificity and RNA activation function (Darbelli and Richard, 2016). Recent research has shown that QKI is downregulated in lung cancer (de Miguel et al., 2016), gastric cancer (Bian et al., 2012), colorectal cancer (Yang et al., 2010), and other tumors and is associated with tumor invasion, metastasis, and prognosis. As a potential tumor suppressor gene, Quaking acts as a pivotal part in endothelial cell differentiation and angiogenesis and is supposed to become an important independent evaluation index in tumor diagnosis and treatment. It has been suggested that QKI may participate in tumorigenesis and development through AS (Zong et al., 2014), cell cycle regulation (Biedermann et al., 2010), and epithelial-to-mesenchymal transition (EMT) (Yang et al., 2016). In this study, we experimentally determined the biological function and underlying regulation mechanism of RNA-binding protein QKI in CC.
We utilized the clinical materials of CC patients in the TCGA database to analyze the difference in QKI expression between CC samples and normal tissues and the relationship between QKI expression level and the clinical staging of patients with CC. The results showed a significantly decreased QKI expression level in CC, and the low expression of QKI contributed to prompt diagnosis of CC. As for why the expression of QKI is obviously decreased in CC, further research is needed, which may be related to DNA methylation (Iwata et al., 2017), gene fusion (Busse et al., 2017), and post-translational modification (Zhang and Feng, 2001). Then, we compared the ASEs between 30 low- and high-QKI expression in CC patients with the ABLas tool and found that in CC clinical samples, QKI extensively regulates the AS of genes that bear basic and important life functions. Therefore, we adopted recombinant plasmids and cell transfection technologies to construct a QKI-OE model in HeLa cells to further explore the effect of QKI on the proliferation and apoptosis of CC cells in vitro and its possible regulatory mechanism. The results witnessed the role of QKI in inhibiting cell proliferation and promoting apoptosis of HeLa cells. It was found that QKI-regulated alternative splicing genes (ASGs) in HeLa cells are mainly enriched in DNA repair, embryonic development, Wnt receptor signaling pathway, and DNA replication. This suggests that QKI may play a critical role in the occurrence and progression of CC through widely regulating the expression and AS of tumor-related genes. It is worth noting that 71% (10/14) of the ASEs were verified by RT-qPCR, which elucidated the validity of RNA-seq data (Fig. 8).
Summary of the research process. First, we compared the QKI expression in cervical cancer and normal tissue samples from the TCGA database. Second, we utilized RNA-seq and functional enrichment analysis to explore the effects of QKI on the occurrence and development of cervical cancer at the transcriptional and post-transcriptional levels. Finally, we verified the identified differentially expressed genes and alternative splicing events by qRT-PCR. Color images are available online.
SR proteins are a class of splicing factors rich in serine and arginine containing an RNA-binding domain (RBD) at the amino-terminal and an RS domain at the carboxyl-terminal. The ability of SR proteins to specifically recognize and bind to pre-mRNA depends on its RBD, while the RS domain involves the localization of SR protein in cells and the interaction between SR proteins and other factors through its phosphorylation. SR proteins regulate AS by combining these two domains with the specific sequence of pre-RNA (Howard and Sanford, 2015; Kędzierska and Piekiełko-Witkowska, 2017). As one of the SR protein family, the serine and arginine-rich splicing factor 10 (SRSF10) with a classical SR protein domain is involved in constitutive and regulated RNA splicing. Studies have found that SRSF10 can control AS of gene transcripts (such as BCLAF1, BRCA1, BCL2L1, CASP8, CHK2, and RBBP8) induced by DNA damage agents to regulate apoptosis, cell cycle transition, and DNA repair (Shkreta et al., 2016; Meng et al., 2019). Zhou et al. (2014) also observed a significant increase in the alternative splicing exon5a inclusion of BCLAL1 transcript in colorectal cancer samples, and the knockdown of this splicing isoform protein significantly inhibited tumor growth. SRSF10 can stimulate the occurrence of exon5a inclusion and has strong tumorigenic characteristics. Higher levels of SRSF10 and exon5a inclusion splicing protein of BCLAL1 are closely related to higher tumor grade. In addition, Liu et al. (2018a) pointed out that SRSF10 regulates the alternate terminator of interleukin-1 receptor accessory protein (IL1RAP) exon 13 to promote the production of the IL1RAP membrane (mIL1RAP) form. SRSF10-mediated mIL1RAP upregulates the expression of CD47 to curb macrophage phagocytosis through activating NF-κB, which is critical in inflammatory processes and tumorigenesis (Liu et al., 2018a). Interestingly, in our data, the overexpression of QKI significantly increased the level of splicing factor SRSF10 in HeLa cells (log2FC = 3.27, p-value = 0.003). And two alternative splicing events (ES and A5SS), located in two relevant genes (RSRC1 and SRSF1), were upregulated and downregulated, respectively, ultimately leading to the inhibition of tumor cell proliferation and the promotion of apoptosis. RSRC1 and SRSF1 are also splicing factors contributing to both constitutive and selective splicing of RNA. Studies have shown that RSRC1 suppresses the proliferation and migration of gastric cancer cells through regulating PTEN expression (Yu et al., 2019). These studies, together with our data, indicate that the impact of many genes on cancer cannot be simply defined as “oncogenic” or “tumor suppressor”. AS is an important reason for this “complexity”. Some splicing isoforms of oncogenes can promote the apoptosis of tumor cells, while some tumor suppressor genes can enhance the migration and invasion of tumor cells after losing a special domain.
Moreover, we observed that detected ASEs regulated by QKI mostly happened in genes associated with transcription regulation and signal transduction, including NFKB1 (nuclear factor of kappa light polypeptide gene enhancer in B cells 1), STAT2 (signal transducer and activator of transcription 2), STAT6 (signal transducer and activator of transcription 6), HRAS (Harvey rat sarcoma viral oncogene homolog), KRAS (Kirsten rat sarcoma viral oncogene homolog), TAF4B (transcription initiation factor TFIID subunit 4B), CEBPZ (CCAAT/enhancer-binding protein zeta), PAXIP1 (PAX interacting protein 1), ELF4 (E74-like factor 4), TCF7L2 (transcription factor 7-like 2), TFAM (transcription factor A, mitochondrial), TADA2A (transcriptional adaptor 2A), USF2 (upstream transcription factor 2), and GTF2H (general transcription factor IIH, polypeptide 1) (Fig. 7 and Supplementary File S15). These genes are generally involved in many vital biological processes, including cell proliferation, differentiation, apoptosis, and immune regulation. This discovery provides a new explanation for the extensive regulation of gene expression by QKI. HRAS, KRAS, and NRAS constitute the most common mutant oncogene family (RAS oncogene family) in human tumors. It has been reported that RAS gene mutations are associated with the occurrence and spread of many tumors, including colorectal cancer (Maffeis et al., 2019), pancreatic cancer (Waters and Der, 2018), lung cancer (Westcott et al., 2015), bladder cancer (Zhang and Zhang, 2015), thyroid tumor (Untch et al., 2018), and myeloid leukemia (You et al., 2018). A variety of transcription variants due to AS have been identified for this gene. In addition, some researchers have also concentrated on the influence of HRAS on the cell cycle progression of CC and proposed that HRAS oncogene can shorten the cell cycle and promote rapid proliferation in CC by reducing the G1 phase and accelerating the G1-S transition of cells (Córdova-Alarcón et al., 2005). Moreover, we found that QKI overexpression significantly upregulates the retention of introns of HRAS (Fig. 7A), which may influence the way QKI regulates CC.
We also observed a significant change in the expression and AS of apoptosis-related genes in our experimental data, including BCL2L11, BCL2L15, BCL6B, BCL11A, and THAP2 in QKI-OE HeLa cells (see details in Supplementary File S15). The protein encoded by BCL2L11 (also called BIM) belongs to the BCL-2 family and contains a Bcl-2 homology domain 3 (BH3) (Kim et al., 2015). Recent studies have shown that it interacts with other members of the BCL-2 family and plays a role in apoptosis activation (Liu et al., 2019). The expression of BCL2L11 can be induced by nerve growth factor (Szegezdi et al., 2008) and forkhead transcription factor (FKHR-L1) (Dijkers et al., 2002), suggesting that this gene may be connected to neuronal and lymphocyte (Wang et al., 2019b) apoptosis. A study on the transgene of the mouse counterpart showed the function of BCL2L11 acting as an essential initiator of apoptosis in thymocyte-negative selection (Bouillet et al., 2002). Furthermore, the research of Kim et al. (2017) demonstrated that BIM level in CC can be applied to predict disease-free survival and overall survival of patients. The higher the BIM, the better the prognosis of patients. Hence, BIM may be an effective chemotherapeutic target for CC in the clinic. THAP2 belongs to the DNA-binding protein family, which is a newly discovered apoptosis-related protein family. It is extensively engaged in important life activities such as cell proliferation, cell cycle, chromosome modification and reconstruction, and transcriptional regulation and is abnormally expressed in various solid tumors. It is a protein that can promote programmed cell death (Morais et al., 2014) and participates in the regulation of tumor invasion, metastasis, and apoptosis (Leite et al., 2011, 2013). A study on bladder cancer cells suggested that THAP2 could interact with prostate apoptosis response-4, playing the role of pro-apoptotic factor, to enhance the sensitivity of cells to serum withdrawal or TNF-α induction and promote the process of programmed cell death (Morais et al., 2014). To sum up, we speculate that downregulation of ES in the BCL2L11 gene and upregulation of A3SS in the THAP2 gene by QKI are the possible mechanisms of promoting apoptosis of CC cells.
Cell cycle disorder is a major pathological mechanism of the malignant tumor. We observed that many genes encoding cyclin (cyclinL1 and cyclinO), GTPase (CDC42 and RASA4), mitogen-activated protein kinase (MAPK), and other proteins related to cell proliferation and division had been alternatively spliced in HeLa cells due to the overexpression of QKI. The expression level of these genes in cells has also changed, which provided a powerful explanation for inhibiting the proliferation of CC cells by QKI. Similarly, other research results also support this theory. Rui-Li Zhang found that overexpression of QKI could prolong the G1 phase and decrease the proportion of the S phase in kidney cancer cells. QKI can bind to the QRE of Ras p21 protein activator 1 (RASA1) mRNA to enhance the stability of RASA1 mRNA, thereby inhibiting the activation of the RAS-MAPK pathway and cell proliferation (Zhang et al., 2016). RASA1 serves as a tumor suppressor gene in many tumors, and the RAS-MAPK pathway acts as a pivotal part in cell proliferation, differentiation, and apoptosis. Therefore, QKI may be involved in regulating G1/S of the cell cycle, inducing cell cycle arrest, and inhibiting cell proliferation through the above effects. In colon epithelium, overexpression of QKI can increase the expression of cyclin-dependent kinase inhibitor p27Kip1, resulting in cell cycle arrest (Yang et al., 2010). In oral squamous cell carcinoma, the low expression of QKI can activate the MAPK signaling pathway and increase the expression of cyclin D1, thus enhancing the proliferation of oral squamous cells. Cyclin D1 plays a key role in the G1/S transition of the cell cycle (Fu and Feng, 2015). Our data analysis and these studies have established QKI's position in regulating the tumor cell cycle.
EMT is regarded as a crucial mechanism for the occurrence, development, invasion, and metastasis of malignant neoplasms (Dongre and Weinberg, 2019). Wnt/β-catenin signaling is one of the important pathways to induce EMT (Gonzalez and Medici, 2014). Our study found that QKI may downregulate the expression of β-catenin standard splice protein by changing the 3′ cleavage site of β-catenin (CTNNB1) transcript. As a key molecule in the Wnt signaling pathway, β-catenin can promote the occurrence and development of CC by activating downstream genes (Li et al., 2018; Liu et al., 2018b). This evidence indicates that QKI can inhibit invasion, metastasis, and EMT of CC by blocking the β-catenin signaling pathway. In a colorectal cancer study, it was also mentioned that overexpression of QKI could enhance the expression level of endogenous β-catenin in the cell membrane, reduce its distribution in cytoplasm and nucleus, and then suppress the transcription activity of β-catenin and block the Wnt signaling pathway, thus inhibiting cancer cell proliferation (Ji et al., 2013). Hypoxi-induced factor-1α (HIF-1α) can promote proliferation, metastasis, EMT, and angiogenesis of various tumors (Masoud and Li, 2015). It has been reported that the knockout of β-catenin can reverse the EMT induced by HIF-1α (Zhao et al., 2011). QKI is significantly associated with HIF-1α. QKI can suppress the expression of HIF-1α in renal cell carcinoma and downregulate the level of target genes GLUT-1, VEGF, and PGK-1 related to glucose metabolism and angiogenesis downstream of HIF-1α (Shi et al., 2020). It is suggested that QKI may play an anticancer role by regulating EMT, which may be related to the inhibition of glucose metabolism and angiogenesis. Wnt/β-catenin signaling pathway and HIF-1α may be involved in this process. In addition, Kim et al. (2019) also found that QKI, as a target of miRNA-200, was involved in the inhibition of EMT and tumor growth. TCF7L2 gene encodes a high mobility group box containing transcription factor, a part of the Wnt/β-catenin pathway, and plays an important part in cell differentiation and growth regulation (Bem et al., 2019). Previously, several studies on CC indicate that the suppression of TCF7L2 expression can effectively prevent the metastasis and progression of CC (Wang and Xia, 2016; Zhou et al., 2017). However, in our data, the overexpression of QKI significantly downregulated the abnormal splicing form of TCF7L2 (CEs) (Fig. 7C) in HeLa cells and finally inhibited the proliferation of tumor cells, suggesting the tumor suppression function of TCF7L2. Besides, in a study of human colorectal cancer, TCF7L2 also showed tumor inhibitory function. Knockout of the nuclear Wnt pathway effector TCF7L2 induces migration and invasion of human colorectal cancer cells (Wenzel et al., 2020). The obvious paradox highlights that the function and underlying regulatory mechanism of TCF7L2 in cancer may need further study.
Finally, it is worth noting a special gene, MUC16, in the overlap analysis between QKI-regulated DEGs and ASGs. MUC16 encodes a transmembrane binding molecule called CA125, which is generally recognized as a specific marker for diagnosing ovarian cancer (Felder et al., 2014). In addition, the level of CA125 in patients with tubal adenocarcinoma, endometrial cancer, CC, pancreatic cancer, colorectal cancer, breast cancer, and lung cancer also increased (Niloff et al., 1984; Integrative Analysis of Lung Cancer Etiology and Risk [INTEGRAL] Consortium for Early Detection of Lung Cancer et al., 2018; Fan et al., 2018; Gao et al., 2018; Li et al., 2019), and the preoperative serum CA125 level of patients can reflect the tumor load and affect the invasion and metastasis of cancer cells. Some studies have suggested that CA125 can bind to mesothelin in a specific way in ovarian cancer to mediate cell attachment between cancer cells and the mesothelial epithelium, thus promoting peritoneal implantation of ovarian cancer (Rump et al., 2004). The research of Chen et al. (2019) proposed that MUC16 induces the movement of p120-catenin from nucleus to cytoplasm and thus activates ras homolog (Rho) GTPases RhoA/CDC42 to regulate the proliferation and migration of ovarian cancer cells. Fan et al. (2018) put forward that the circulating regulatory T cell subset (Treg) is an independent prognostic indicator of pancreatic cancer. In pancreatic cancer, MUC16 C-terminal (MUC16c) expression is significantly increased, which is involved in the regulation of IL-6 expression and secretion through PI3K/AKT pathway. The tumor-derived IL-6 can promote the expression of Foxp3 and the differentiation of Treg, which finally affect the prognosis of pancreatic cancer patients. Our results indicated that the level of MUC16 was significantly downregulated in QKI-OE HeLa cells (log2FC = −1.05, p-value <0.001). A novel ASE, ES, was identified in the MUC16 transcript (Fig. 7B and Supplementary File S15), implying that QKI's coordinated regulation of AS (post-transcriptional level) and gene expression (transcriptional level) of MUC16 in CC may be a powerful explanation for its role as a tumor suppressor.
Conclusion
Generally, we successfully verified the regulation effect of QKI on AS through RNA-seq technology. In HeLa cells, QKI suppresses the proliferation and induces apoptosis of tumor cells by regulating the AS of a series of genes related to cancer occurrence and progression, and this corresponds to its reported function as a tumor suppressor. Our results emphasize that QKI can regulate the expression of downstream targets through AS and participate in multiple biological processes, including cell cycle modulation, cell proliferation, angiogenesis, apoptosis, EMT, and immune regulation. QKI is of great significance in suppressing malignant tumors and may become a new biomarker for the diagnosis, treatment, and evaluation of malignant tumors in the future. The potential downstream molecular mechanism of QKI protein in inhibiting cancer provides a direction for further research.
Authors' Contribution
N.J. provided article design. Y.L. performed experimental and data analysis. Y.L. and P.G. drafted the article. K.Z. and H.L. had assistance with the article review. All authors participated in the correction and approval of the final article.
Footnotes
Acknowledgments
The authors are especially grateful to Dr. Chao Cheng and ABLife, Inc., for the language polishing and technical consultation.
Disclosure Statement
The authors declare that they have no financial or other conflicts of interest.
Funding Information
No funding was received.
Supplementary Material
Supplementary Figure S1
Supplementary File S1
Supplementary File S2
Supplementary File S3
Supplementary File S4
Supplementary File S5
Supplementary File S6
Supplementary File S7
Supplementary File S8
Supplementary File S9
Supplementary File S10
Supplementary File S11
Supplementary File S12
Supplementary File S13
Supplementary File S14
Supplementary File S15
References
Supplementary Material
Please find the following supplemental material available below.
For Open Access articles published under a Creative Commons License, all supplemental material carries the same license as the article it is associated with.
For non-Open Access articles published, all supplemental material carries a non-exclusive license, and permission requests for re-use of supplemental material or any part of supplemental material shall be sent directly to the copyright owner as specified in the copyright notice associated with the article.