
Abstract
Dysfunction of bone marrow mesenchymal stem cells (BMSCs) is recognized critical in bone deteriorations of osteoporosis. However, the specific mechanisms that determine the fate of BMSCs remain elusive. MicroRNA-133a (miR-133a), a highly conserved microRNA, was investigated under both in vitro and in vivo conditions. In the in vitro study, cell proliferation, cell apoptosis, and osteoblast/adipocyte differentiation of BMSCs as a result of overexpression or knockdown of miR-133a was investigated. In the in vivo study, the ovariectomy (OVX) model was applied on mice, with further treatment of the models with BMSC-specific miR-133a antagomir through femur intramedullary injection. Microcomputed tomography scanning and histological analysis of the proximal and middle femur were performed to evaluate the morphological changes. The results revealed that overexpression of miR-133a suppressed cell proliferation, cell viability, and osteoblast differentiation of BMSCs, but increased adipocyte differentiation. We also found that FGFR1, an important upstream regulator of mitogen-activated protein kinase/extracellular signal-regulated kinase (MAPK/ERK) signal pathway, was a major target of miR-133a. We also recorded that BMSC-specific knockdown of miR-133a attenuates bone loss in OVX mice. Our study suggested that miR-133a played an important role in maintaining the viability and balance between osteoblast and adipocyte differentiation of BMSCs through the MAPK/ERK signaling pathway by targeting FGFR1.
Introduction
Osteoporosis is a multifactorial disorder characterized by skeletal fragility and microarchitectural deterioration (Black and Rosen, 2016). The disruption of the balance between osteoblastic formation and osteoclastic resorption and dysfunction of the bone marrow mesenchymal stem cells (BMSCs) leads to bone deterioration as well as plays a key role in the pathogenesis of the disease (Zheng et al., 2020).
Similar to how osteoblasts form the bone tissues, BMSC plays a critical role in bone remodeling, by directly affecting bone formation and bone resorption (Zhang et al., 2016; Meng et al., 2019). Recent studies have revealed that several risk factors are associated with elderly people, such as more senescent cells, slower cell expansion, increased cell apoptosis, and decreased osteoblast differentiation of BMSCs (Chen, 2004; Zhou et al., 2008). Past studies have indicated that osteoporosis is associated with an increase in the number of adipocyte cells in the BMSCs and a decrease in the number of osteoblast cells (Li et al., 2015); bone mass and density can be enhanced by regulating the balance between osteogenesis and adipogenesis (Jiang et al., 2018).
Several studies that investigated the role of microRNA-133a (miR-133a) in bone metabolism revealed that miR-133a is upregulated in mice after ovariectomy and in mice with reduced trabecular thickness (
All of the above-mentioned studies indicated that miR-133a played a key role in the occurrence of osteoporosis, especially in terms of the abnormality associated with the differentiation of BMSCs. However, the underlying mechanism remains unclear. Therefore, this study purports to investigate the effects of miR-133a on BMSCs and the associated mechanisms.
Materials and Methods
Cell culture and transfection
Human BMSCs obtained from American type culture collection (ATCC, PSC-500-012) were cultured in HyClone DMEM containing 10% FBS (Gibco), penicillin G (100 U/mL; Gibco), and streptomycin (100 μg/mL; Gibco) at 37°C in a humidified atmosphere of 5% CO2. Passage was performed when the adherent cultured cells obtained 80–90% confluency; BMSCs at passage 3–6 were used in the in vitro experiments.
To modulate the expression of miR-133a in BMSCs, cells were transfected with miR-133a agomir (RIBOBIO, Guangzhou, China) or miR-133a antagomir (RIBOBIO) and their negative controls according to the manufacturer's instructions. After transfection, BMSCs were cultured in fresh DMEM medium for further experiments.
Cell proliferation and apoptosis
To detect the effect of miR-133a on BMSC proliferation, BMSCs (4000/well) were cultured in 96-well plates. After 24 h (day 1), 48 h (day 2), 72 h (day 3), 96 h (day 4), and 120 h (day 5) incubation, respectively, the proliferation culture medium containing FBS was discarded and then 10 μL of cell counting kit-8 (CCK-8) (Beyotime; Shanghai, China) solution and 90 μL of DMEM were added into the wells, followed by incubation for another 2 h. The A values at 450 nm were recorded with the help of a microplate A reader.
Cell apoptosis was induced by using the serum-starved culture method and was detected with the help of Annexin V-FITC-PI Apoptosis Detection Kit (Beyotime). BMSCs, after being cultured for 96 h, were harvested and resuspended in 200 μL of Annexin V-FITC in conjunction with 10 μL of propidium iodide. Finally, flow cytometry was used to detect and quantify apoptotic and dead cells.
Osteoblast differentiation and adipocyte differentiation
The differentiation in BMSCs was induced by using osteogenic induction medium (full culture medium containing 10−2 M β-sodium glycerophosphate, 50 μg/mL
After being subjected to osteogenic induction for 21 days and adipogenic induction for 14 days, the BMSCs were stained with Alizarin Red S (Beyotime) or oil red (Beyotime), then the osteoblastic mineralization or mature adipocytes was assessed using an inverted phase-contrast microscope. Quantification was performed using Image Pro Plus 6.0 software from at least five randomly selected microscopic fields, the relative area of mineralized matrix was calculated as the area of mineralized matrix/total area, and the relative area of adipocytes was calculated as the area of adipocytes/total area.
Western blotting
To detect the expression of biomarker proteins associated with osteogenesis and adipogenesis, total cell lysates were collected, and a BCA Kit (Beyotime Biotechnology) was used to determine the protein concentration. After denaturation, a 30-μg sample protein was separated using sodium dodecyl sulfate-polyacrylamide gel electrophoresis and was then transferred to a polyvinylidene fluoride membrane (Millipore).
The membrane was blocked with 5% BSA and labeled with primary antibodies to RunX2 (ab23981; Abcam), ALP (ab83259; Abcam), ERK1/2 (4695; CST), p-ERK1/2 (4376; CST), ap2 (ab92501; Abcam), peroxisome proliferator-activated receptor gamma (PPARγ) (ab209350; Abcam), FGFR1 (ab31324; Abcam), and GAPDH (ab181602; Abcam) at a concentration of 1:1000 overnight, followed by immersion in Horseradish peroxidase (HRP)-conjugated secondary antibodies (31160, 31210; Thermo Pierce) for 1 h. The blotted bands were analyzed using an electrochemiluminescence kit (Santa Cruz Biotechnology, Santa Cruz, CA) and visualized using X-ray films.
Quantitative real-time PCR analysis
Total RNA was extracted from the BMSCs using TRIzol reagent (Invitrogen) and analyzed using the NanoDrop Spectrophotometer. Next, reverse transcription was performed using 1 μg of total RNA and EasyScript one-step gDNA Removal and cDNA Synthesis Supermix (TransGen Biotech, Beijing, China). Quantitative real-time PCR (qRT-PCR) was performed to assess the expression of target mRNAs. The reaction conditions during the process were as follows: 95°C temperature was maintained for 30 s; maintained at 95°C for 5 s; and finally at 60°C for 30 s in all 40 cycles. The sequences of forward and reverse primers have been listed in Table 1.
Primer Sequences in the Quantitative Real-Time Polymerase Chain Reaction Analysis
PPARγ, peroxisome proliferator-activated receptor gamma.
Dual-luciferase reporter assay
Target genes of miR-133a were predicated using TargetScan (
The BMSCs (2*104/well) were inoculated in 24-well plates and transfected with WT or mutant pmirGLO construct, miR-133a mimics or negative control, and pRL-TK Renilla luciferase plasmid (Promega) using the X-treme Gene HP DNA Transfection Reagent (ROCHE, Switzerland). The firefly and Renilla luciferase activity was tested using the dual-luciferase reporter assay system (Promega). The luciferase activity was normalized to Renilla luciferase activity that represented the expression of FGFR1.
Construction of nanocomplex containing BMSC-specific aptamer
The construction of BMSC-specific aptamer delivery system was performed as described previously (Li et al., 2015; Zhao et al., 2011). The polyethyleneimine (PEI)-citrate core structure (nanocore) was constructed by mixing one part of PEI solution (100 μg/mL; pH 6.0) and six parts of sodium citrate (4.2 μM). Three parts of synthetic BMSC aptamers (50 nM) and mouse miR-133a antagomir (100 nM, synthesized by RiboBio Co.) or the negative control (100 nM) was added to the nanocore to assemble the nanocomplex after 5 min of reaction.
Animal models and treatment
For this study, 24 C57BL/6j mice were randomly divided into four groups: (1) control group, mice were ovariectomy (OVX) sham operated; (2) OVX group (bilaterally ovariectomized mice); (3) OVX+Antagomir-133a group (mice were bilaterally ovariectomized and treated with BMSC-specific miR-133a antagomir); and (4) Antagomir NC group (mice were bilaterally ovariectomized and treated with BMSC-specific miR-133a antagomir negative control).
Next, the mice were either treated with 40 μL of miR-133a antagomir nanocomplex or the negative control nanocomplex or PBS by injecting it into bilateral femur medullary cavity every 2 weeks. Finally, the mortality rates in the subgroups were 1/6 in the control group, 1/6 in the OVX group, 1/6 in the OVX+Antagomir-133a group, and 2/6 in the Antagomir NC group. All femurs of these mice were obtained 8 weeks after the operation. All experiments concerning animals were approved by the Institutional Animal Care and Use Committee of Anhui Medical University (LLSC20190262).
Microcomputed tomography scanning and analysis
To evaluate the femoral morphology in these samples, four left and four right femurs in each group were treated with 4% paraformaldehyde for 24 h and then scanned using a microcomputed tomography (microCT) scanner at a resolution of 9 μm. The region of interest (ROI) of trabecular bone was selected, which was the area (2–2.9 mm) below the femoral head and was analyzed for bone mineral density (
Histochemical analysis
To further evaluate the microstructure of these models, the left femurs were decalcified by using 10% ethylenediaminetetraacetic acid for 2 weeks after microCT scanning, and then embedded with paraffin. Four proximal femurs in each group were cut into 5-μm thickness sections in the coronal plane and labeled with hematoxylin and eosin staining (two slices of each femur) and immunohistochemical staining for osteocalcin (two slices of each femur) as described previously (Lin et al., 2019).
Statistical analysis
SPSS 17.0 was used for statistical analysis of the data. All data were expressed in terms of means and standard deviations. One-way analysis of variance (ANOVA) was employed along with Tukey's post hoc test. All experiments were repeated at least thrice. p < 0.05 was indicative of significant difference.
Results
Inhibition of BMSC proliferation and promotion of BMSC apoptosis by miR-133a
To regulate the expression of miR-133a in BMSCs, the BMSCs were transfected with miR-133a agomir or miR-133a antagomir, and the qRT-PCR results revealed that the BMSCs showed significant upregulation when transfected with miR-133a agomir and showed downregulation when transfected with miR-133a antagomir (Fig. 1A). The transfected BMSCs were then cultured with full medium to study their proliferation ability. The CCK-8 results indicated that the overexpression of miR-133a inhibited BMSC proliferation, whereas knockdown of miR-133a promoted it significantly (Fig. 1B).
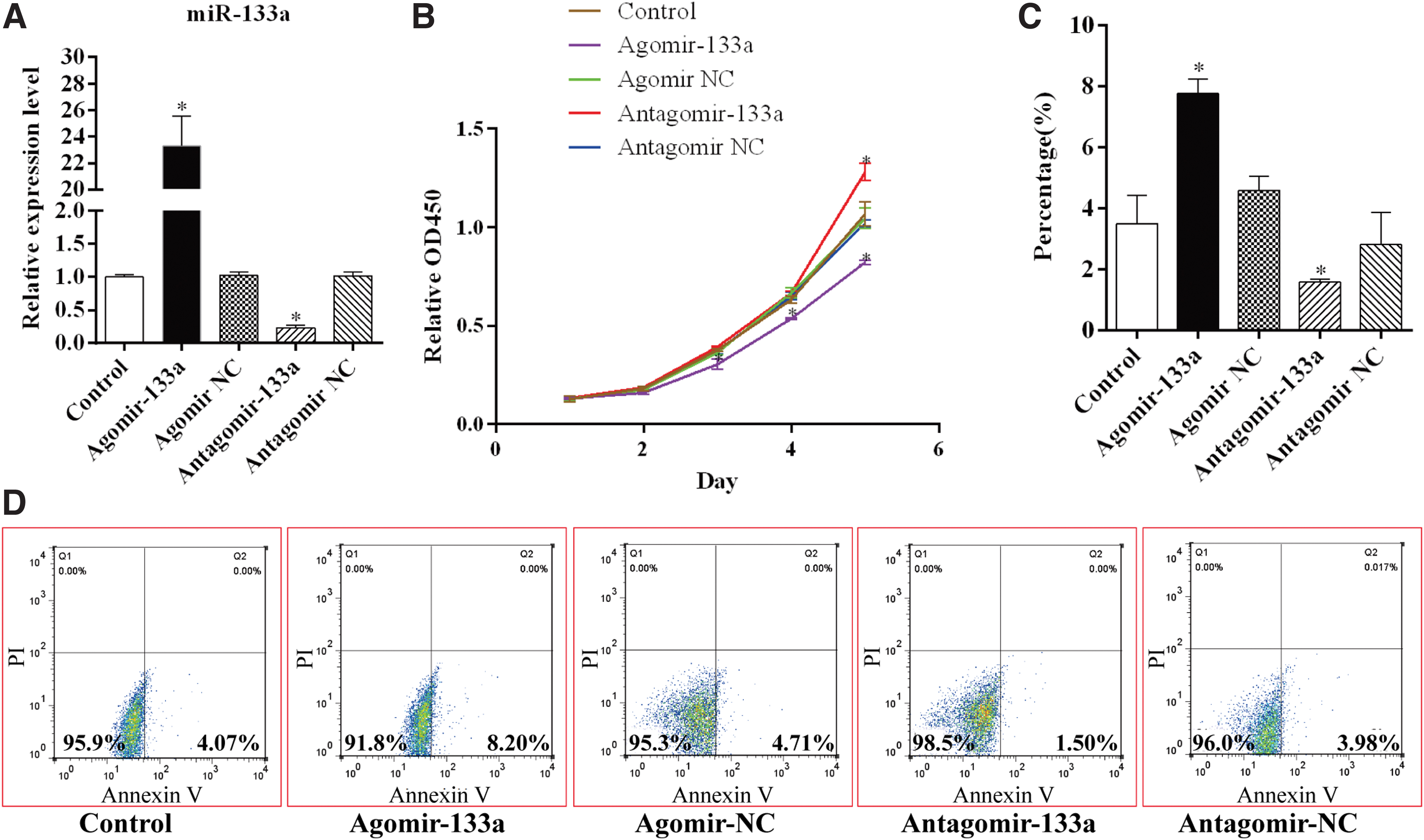
MiR-133a inhibits cell proliferation and promotes cell apoptosis of BMSC.
Further investigation of BMSC apoptosis was performed using FBS-free medium, and the results of the flow cytometric analysis indicated that the overexpression of miR-133a increased the chances of early apoptosis in BMSCs due to Annexin-V positive and propidium iodide (PI) negative staining. In contrast, the downregulation of miR-133a inhibited BMSC apoptosis to a very large extent (Fig. 1C, D).
Inhibition of osteoblast differentiation in BMSCs by miR-133a
To explore the effect of miR-133a on BMSC osteoblast differentiation, BMSCs with miR-133a agomir or miR-133a antagomir transfection were induced for osteoblast differentiation for 48 h. The results of western blotting analysis revealed that the expression of two osteoblast differentiation markers (Runx2 and ALP protein) were lower in the miR-133a agomir group than that in the control groups, whereas the knockdown of miR-133 caused significant upregulation in the expression of Runx2 and ALP (Fig. 2A–C). Moreover, the mRNA level of Runx2 and ALP also showed downregulation due to the overexpression of miR-133a (Fig. 2D, E).
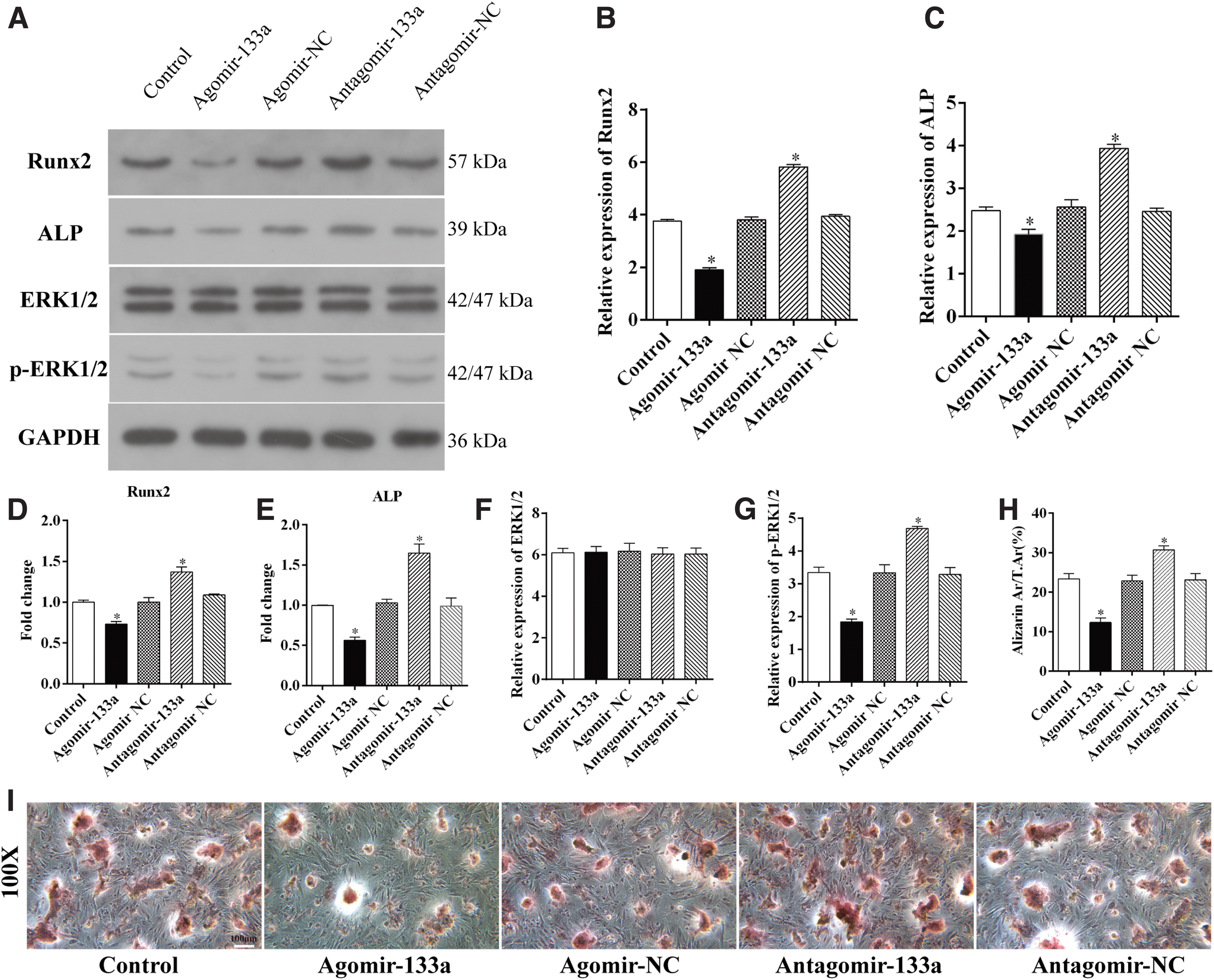
MiR-133a suppresses osteoblast differentiation of BMSCs.
Use of Alizarin red staining further demonstrated the inhibiting effect of miR-133a on BMSC osteoblast differentiation. After osteoblastic induction was performed for 21 days, BMSCs with overexpressed miR-133a demonstrated fewer calcium nodules when compared with the control group, whereas the knockdown of miR-133a caused an increase in the calcium nodules in BMSCs (Fig. 2H, I).
To further investigate the mechanism underlying osteoblast differentiation, the expression of ERK and p-ERK, which are the key proteins in the mitogen-activated protein kinase/extracellular signal-regulated kinase (MAPK/ERK) signaling pathway, were analyzed by using the western blot technique. The results revealed that the overexpression of miR-133 inhibited the phosphorylation of ERK1/2 and the knockdown of miR-133 caused upregulation of the level of p-ERK1/2 (Fig. 2A, F, G), indicating that miR-133a is responsible for the regulation of osteogenic differentiation of BMSCs, possibly through the regulation of the MAPK/ERK signaling pathway.
Promotion of adipocyte differentiation of BMSCs by miR-133a
As described earlier, the overexpression or knockdown of miR-133a in BMSCs was induced by using agomir or antagomir. Then, BMSCs were cultured in adipogenesis induction medium for 48 h, and the expression of aP2 and PPARγ was analyzed by western blotting and qRT-PCR techniques. The results revealed that both protein and mRNA expression was upregulated by miR-133a. In contrast, the knockdown of miR-133a downregulated the expression of aP2 and PPARγ significantly, indicating regulation of adipocyte differentiation-related proteins by miR-133a (Fig. 3A–E).

MiR-133a promotes adipocyte differentiation of BMSC.
Oil red staining indicated a significant decrease in the number and area of adipocytes in the case of miR-133a antagomir-transfected BMSCs and increase in the case of miR-133a agomir transfected BMSCs after adipogenesis differentiation for 14 days, indicating that the overexpression of miR-133a facilitated lipid droplet formation and knockdown of miR-133a, attenuated adipocyte differentiation of BMSCs (Fig. 3H, I). Phosphorylation of ERK1/2 was also inhibited by miR-133a in BMSCs cultured in adipogenesis induction medium, indicating that the MAPK/ERK signaling pathway was involved in the regulation of BMSC adipocyte differentiation by miR-133a (Fig. 3A, F, G).
miR-133a directly targets FGFR1
To explore the mechanism of inactivation of MAPK/ERK signaling as a result of overexpression of miR-133a, the miR-133a targets were predicted using TargetScan. It was discovered that the 3′-UTR of FGFR1mRNA, an upstream regulator of MAPK/ERK signaling, matched seven nucleotides of miR-133a at two sites, from 255 to 261 and from 530 to 536, indicating that miR-133a modulates the expression of FGFR1. Thus, the expression of FGFR1 in BMSCs was analyzed using miR-133a. The results showed that miR-133a downregulated the level of FGFR1 protein and mRNA significantly, whereas BMSCs along with miR-133a silence showed a higher expression of FGFR1 (Fig. 4A–C).
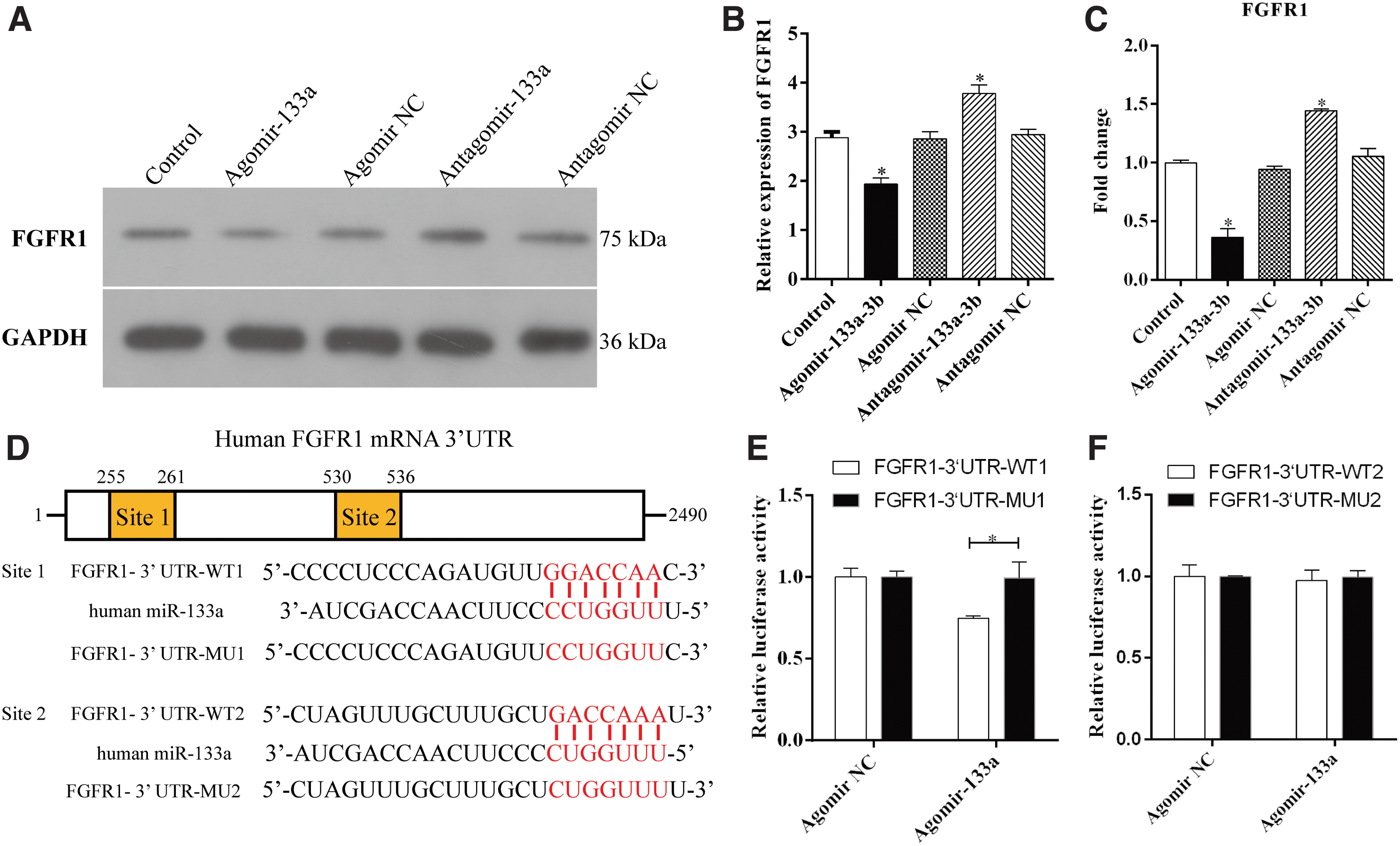
MiR-133a directly targets FGFR1.
This was followed by cloning of segments of human FGFR1 mRNA 3′-UTR containing the two predicted binding sites of miR-133a (FGFR1-3′ UTR-WT1 and FGFR1-3′ UTR-WT2) or two different mutated sites (FGFR1-3′ UTR-MU1 and FGFR1-3′ UTR-MU2) into pmirGLO vector separately. The dual-luciferase reporter assay illustrated that the miR-133a ectopic expression significantly reduced the luciferase activity of the FGFR1-3′ UTR-WT1 reporter plasmids, but not that of the FGFR1-3′ UTR-MU1 reporters. However, the miR-133a expression exerted no effect on the luciferase activity of both FGFR1-3′ UTR-WT2 and FGFR1-3′ UTR-MU2 reporter plasmids (Fig. 4D–F), indicating that FGFR1 is the target of miR-133a in humans and the target sites were the seven nucleotides (255–261), but not the nucleotides (530–536).
Targeted silencing of miR-133a attenuates bone loss in OVX mice
To investigate the effect of BMSC-specific silence of miR-133a on bone metabolism, BMSC-specific aptamer delivery system was constructed, and then miR-133a antagomir nanocomplex and the negative control were injected into the femoral cavity of the OVX mice. MicroCT analysis on the proximal femurs showed that the mean
In addition, the other parameters, including Tb. BV/TV, Tb. Th and Tb. N, were lower in OVX mice, but increased as a result of knockdown of miR-133a. The Tb.Sp and Tb.Pf, which are the parameters representing the degree of trabecular deterioration, were higher in the OVX mice relative to the controls, but decreased as a result of knockdown of miR-133a (Fig. 5A–G), indicating that the inhibition of miR-133a attenuated bone loss in OVX mice. Similar to the microCT results, histological staining showed sparser trabecula and fewer osteocalcin-positive osteoblasts in the proximal femur of OVX mice as compared with that of the control cases. Notably, the number of trabecula and osteocalcin-positive osteoblasts increased in mice treated with miR-133a antagomir nanocomplex (Fig. 5H–I).
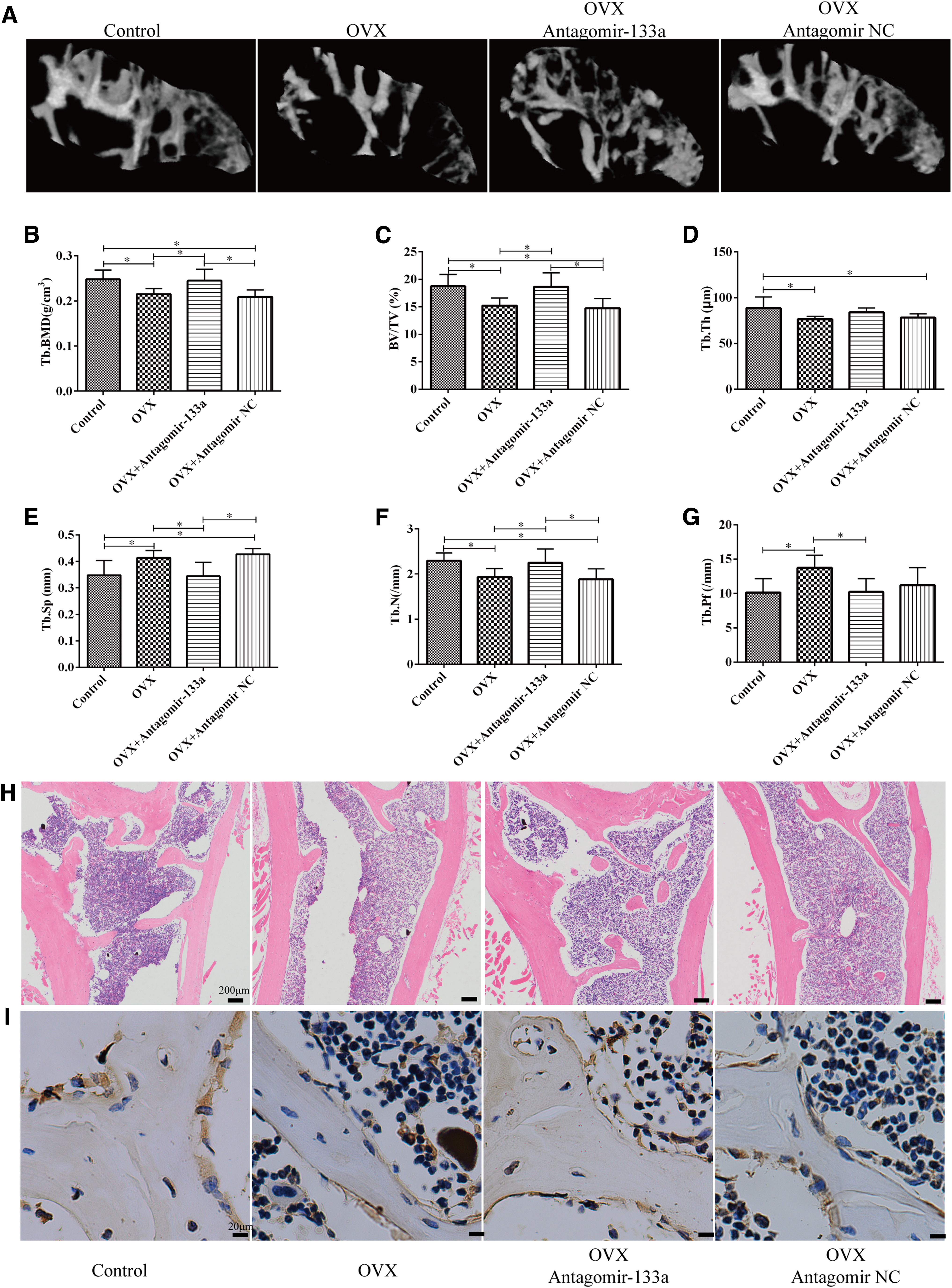
Targeted silencing of miR-133a attenuates trabecular deterioration of proximal femur in OVX mice.
Furthermore, the analysis of the microarchitecture of cortical bone in the middle femur was performed. The results revealed that the mean BV and
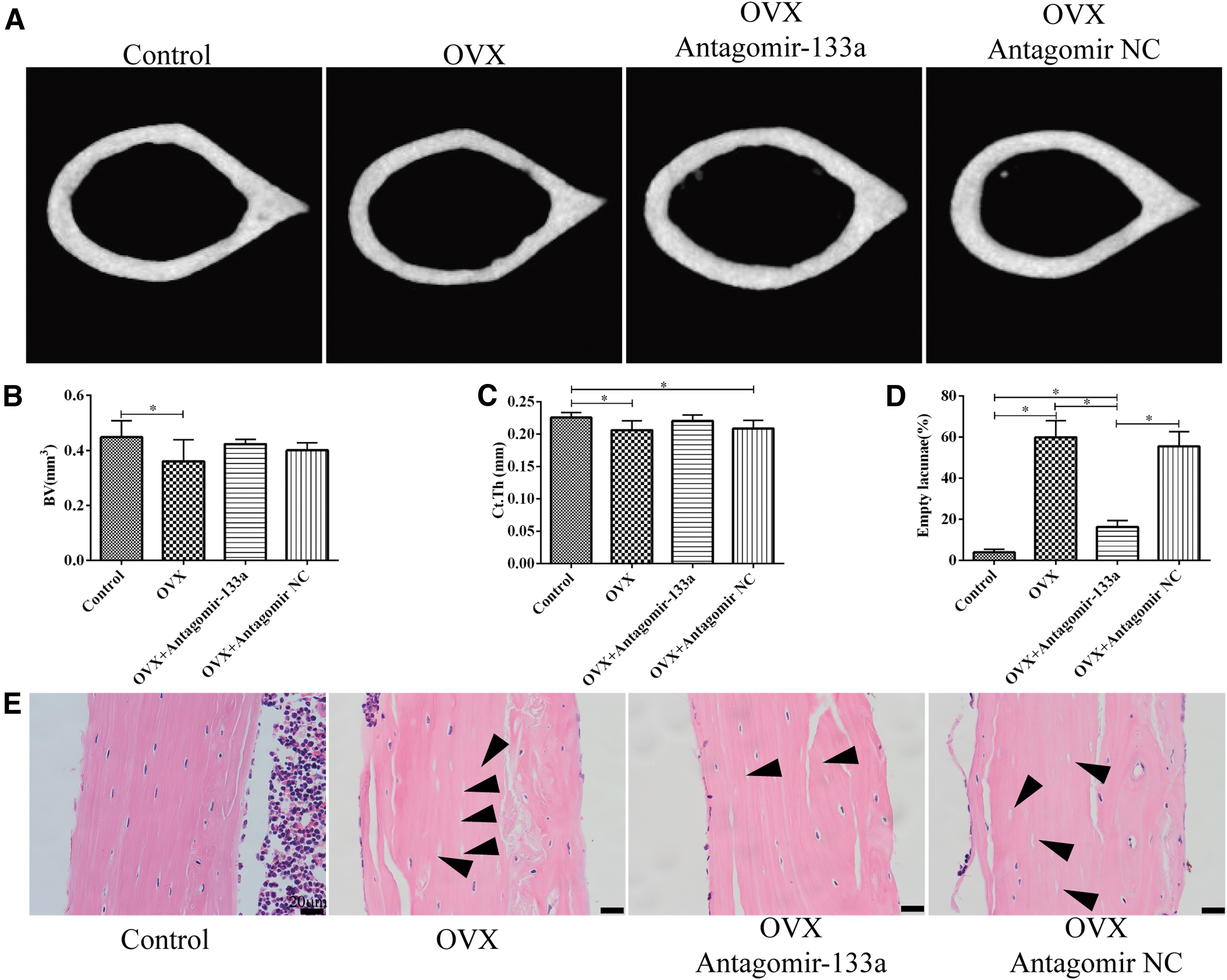
Targeted silencing of miR-133a ameliorates cortical microarchitecture of femur in OVX mice.
Discussion
This study provides detailed observations regarding the effect of miR-133a on the activity of BMSCs. We found that miR-133a negatively regulated the proliferation and viability of BMSCs; the ectopic overexpression of miR-133a suppressed the osteoblast differentiation and stimulated the adipocyte differentiation of BMSCs; whereas inhibition of miR-133a expression blocked this effect and had a countereffect on bone loss in OVX mice. We also concluded that the possible molecular mechanism underlying the effect of miR-133a on BMSC activity may be the inactivation of MAPK/ERK signaling by targeting FGFR1.
The MAPKs play important roles in cellular response to cytokines, growth factors, or environmental stress (Fu et al., 2008). Previous studies have reported at least four MAPK family members, including ERK, JNK, p38, and ERK5, involved in bone metabolism. The MAPK/ERK pathway is involved in BMSC proliferation, migration, apoptosis, and activation of ERK signaling promotes BMSC proliferation, migration, and increases cell survival against apoptosis (Jeong et al., 2017; Liang et al., 2019). Moreover, it was observed that a sustained ERK activation occurred during osteoblast differentiation of BMSCs, and inhibition of the ERK signaling pathway blocked osteoblast differentiation and promoted adipocyte differentiation in BMSCs (Ge et al., 2016a, 2016b).
In addition, studies have also reported the involvement of MAPK/ERK signaling pathway in the process of BMSC differentiation regulated by miRNAs, such as miR-205 (Hu et al., 2015), miR-21 (Mei et al., 2013), and miR-16 (Qi et al., 2020). The results of the study were also consistent with the earlier reported findings that suggested that miR-133a regulated the balance of osteogenic and adipocyte differentiation in BMSCs by modulating the MAPK/ERK activity. Furthermore, the proliferation ability and viability were also affected by the ERK phosphorylation regulated by miR-133a.
As reported previously, the differentiation of BMSC into osteoblast or adipocyte was controlled by the relative activity of RunX2 and PPAR-γ (Hong, 2005). The result of the study revealed that PPAR-γ is an important marker of adipocyte differentiation and lipid metabolism, and the suppression of PPAR-γ function leads to reduced adipocyte differentiation in mesenchymal stem cells. This finding is consistent with the findings reported elsewhere (Hong, 2005). Runx2 is another important transcription factor responsible for the control of differentiation in BMSC through manipulation of Runx2 in cell cultures and in animal models (Ducy et al., 1997).
Both RunX2 and PPAR-γ were regulated by MAPK/ERK phosphorylation. MAPK-dependent phosphorylation of PPAR-γ at S112 inhibits both ligand-dependent and ligand-independent activities and blocks adipogenesis (Ge et al., 2016a, 2016b). P-ERK translocates from perinuclear to nuclear sites for phosphorylation of at least four serine residues of RUNX2, which were previous to bounding with promoter regions of target genes, resulting in increased transcriptional activity and stimulation of osteogenesis.
Furthermore, RunX2 has been considered to be a target of miR-133a in BMP-2 induced osteoblast differentiation of mesenchymal cells, and it directly binds to the 3′-UTR of RunX2 mRNA to block Runx2 protein translation (Li et al., 2008). In this study, the expression and activity of RunX2 and PPAR-γ were both regulated by miR-133a, accompanied by the tendency of BMSC differentiation and variation of ERK phosphorylation, indicating that miR-133a modulates BMSCs differentiation partly by the inactivation of the MAPK/ERK signaling pathway in addition to RunX2.
Fibroblast growth factor (FGF)/FGFR signaling plays an important role in skeletal development through the stimulation of multiple pathways, including Ras-MAP kinase and PI-3 Kinase/AKT pathways (Du et al., 2012). As the main regulator of MAPK/ERK signaling (Suzuki et al., 2000; Lu et al., 2009), the expression of FGFR1 in mesenchymal cells has been well characterized (Maruyama et al., 2010; Du et al., 2012). It has been reported that FGFR1 signaling stimulates proliferation and has a positive influence on the regulation of cell viability in human mesenchymal stem cells (Dombrowski et al., 2013, Ornitz and Marie, 2015). Moreover, some studies have indicated that inhibiting or silencing FGFR1 using specific inhibitor or specific shRNAs abrogates osteoblast gene expression and matrix mineralization in mesenchymal stem cells (Hamidouche et al., 2010, Kim et al., 2012, Zhang et al., 2015).
This study illustrated the relationship between FGFR1 and miR-133a in BMSCs. MiR-133a binds directly with 3′-UTR of FGFR1 mRNA and decreases its protein level, followed by the inactivation of FGF/FGFR1 downstream signaling, leading the disruption of BMSC viability and its differentiation ability.
Currently, various medications and therapeutic options has been established for the treatment of osteoporosis, and pharmacological treatment that could be divided into antiresorptive agents and anabolic agents remain strongly recommended. Nevertheless, understanding the detailed mechanism underlying osteoporosis makes novel targets and novel approach feasible. Stem cell-based and target gene regulation therapies have been conducted in osteoporotic animal models and turned out to be very promising (Noh et al., 2020).
The efficiency of MSCs has been investigated extensively both in basic and clinical experiments during the past decades, and according to their sources and characteristics, MSCs could be divided into BMSCs, dental-derived MSCs (Bartold et al., 2019, Tatullo et al., 2019), adipose-derived MSCs, perivascular stem cells, induced pluripotent stem cells and genetically modified MSCs (Jin and Lee, 2018). These populations of MSCs are all potential candidates for bone regeneration by stimulation of osteogenesis.
In this study, we explored the specific mechanism of miR-133a regulating BMSCs, miR-133a has also been reported to be involved in the osteogenic differentiation of other populations of MSCs, especially in dental-derived MSCs. Du et al. (2019) revealed downregulated expression of miR-133a in the process of osteogenesis in rat dental follicle cells, Iaculli et al. discovered that miR-133a was downregulated in dental pulp stem cells grown on the titanium disks, which showed an earlier differentiation process compared with the control (Iaculli et al., 2017), and the expression of miR-133a was also inhibited in the periodontal ligament stem cells (PDLSCs) cultured with ibandronate (Zhou et al., 2011). However, BMSCs have been the most widely studied and utilized in clinical settings, including spinal fusion surgery, nonunion, and femoral head osteonecrosis (Jin and Lee, 2018).
Targeted miRNA therapy in MSCs for bone regeneration has been studied for a long time, Deng discovered that anti-miR-31-expressing BMSCs exhibited good biocompatibility and a high regeneration rate within in vivo bone defects (Deng et al., 2014) and Xie indicated that miR-135-modified adipose-derived MSCs effectively repaired critical-sized bone defects (Xie et al., 2016). However, the strategies of pre-transduction of stem cells with viral pre-miRNAs or miRNA inhibitors were limited in clinical translation considering the insertional mutagenesis risk and adverse immune responses associated with viruses (Castano et al., 2020). Then, nonviral technologies protecting miRNA and enhancing delivery has been developed, including collagen-nanohydroxyapatite scaffold (Castaño et al., 2015) and PEI-citrate core structure used in our study.
Although these nonviral technologies decrease risks from viruses or plasmids, miRNAs are capable of targeting multiple proteins with potential complications. To our knowledge, miR-133a is critical for skeletal muscle and cardiac development as a potentially powerful biomarker of acute myocardial infarction (Xiao et al., 2019, Xie et al., 2018), miR-133a also plays an important role in brown fat differentiation and adipocyte browning (Liu et al., 2013). Systemic or local knockdown of miR-133a possibly affects other tissue and cell lines.
Aptamers are single-stranded nucleic acid molecules that bind to targets through folding into a three-dimensional structure with high affinity and selectivity, aptamer-conjugated nanocore has been utilized to deliver antagomiR-188 into mice BMSCs to overcome age-related osteoporosis. In this study, we used the aptamer-conjugated nanocore system to silence miR-133a expression in BMSCs of the femoral cavity without interference with other cell lines, and results showed that BMSC-specific silence of miR-133a effectively attenuated bone loss in OVX mice.
In addition, bone formation and regeneration involve very complex and highly regulated cellular and molecular processes (Bartold et al., 2019), this study focused only on the effect of miR-133a on BMSCs without exploring the effects on other cells involving in bone metabolism. Studies showed that miR-133a inhibits the expression of osteoblast differentiation-associated markers in osteoblast-like cells (Zhang et al., 2018), miR-133a was also upregulated during osteoclastogenesis and promoted RANKL-induced differentiation of RAW264.7 and THP-1 cells into osteoclasts (Li et al., 2018). Moreover, the potential targets of miR-133a, including SLC39A1 (Wang et al., 2017) and PRDM16 (Liu et al., 2013), may also be involved in the regulation of miR-133a on bone metabolism. Therefore, BMSC was not the only source of bone formation to overcome osteoporosis, miR-133a targeted treatment still needs further study except BMSC.
Conclusion
In conclusion, the findings of this study suggested that miR-133a regulates the viability and the balance between osteoblast and adipocyte differentiation of BMSCs through the inhibition of the MAPK/ERK signaling pathway by targeting FGFR1. Local knockdown of miR-133a may be a promising method for treating patients suffering from osteoporosis.
Footnotes
Disclosure Statement
No competing financial interests exist.
Funding Information
This study was funded by the National Natural Science Foundation of China (no. 81702134) and Anhui Medical University School Foundation (2020xkj183).