
Abstract
Skeletal muscle has great plasticity. An increase in protein degradation can cause muscle atrophy. Atrogin-1 and muscle ring finger-1 (MuRF1) are dramatically upregulated in various muscle atrophy. Inhibition of Atrogin-1 and MuRF1 protects against muscle atrophy. MiR-29 plays an important regulatory role in skeletal muscle development. However, the function of miR-29 in skeletal muscle protein metabolism is not clear. To investigate the function of miR-29, we generated miR-29 knockout mice and the miR-29ab1 cluster overexpression mice. The disruption of miR-29 led to severe atrophy of skeletal muscle during puberty, and the muscle-specific overexpression of the miR-29ab1 cluster protected against denervation-induced and fasting-induced muscle atrophy. Furthermore, the overexpression of miR-29a, b mimics in myotubes resisted the muscle atrophy. MuRF1 was the direct target gene of miR-29a, b. These results demonstrate that miR-29ab1 cluster plays a critical role in the maintenance of skeletal muscle. MiR-29ab1 cluster is the excellent inhibitor of MuRF1, ultimately indicating that miR-29ab1 cluster is good therapeutic molecule candidate for adulthood.
Introduction
Skeletal muscle size is related to protein synthesis and protein degradation. Increased anabolism causes hypertrophy and increased catabolism causes atrophy (Latres et al., 2005). Skeletal muscle atrophy develops in fasting, AIDS, cancer, disuse, or aging (Day et al., 2002; Liu et al., 2017). Certain genes are increased and can regulate the physical and pathological progression of skeletal muscle atrophy (Bonaldo and Sandri, 2013). Ubiquitin-mediated proteolysis has been shown to play a key role in muscle protein degradation (Bodine and Baehr, 2014).
Atrogin-1 and muscle ring finger-1 (MuRF1) are the E3 ubiquitin ligases and dramatically upregulated in various muscle atrophy (Lecker et al., 2004; Glass, 2010; Liu et al., 2017). They have been shown to target and ubiquitinate certain sarcomeric proteins and are involved in the muscle ubiquitin proteasome pathway (Lecker et al., 2004; Clarke et al., 2007). In addition, MuRF1 participates in the inhibition of muscle protein synthetic pathways (Baehr et al., 2011). MuRF1 or Atrogin-1 knockout mice show a reduced muscle loss ratio during atrophy (Bodine et al., 2001; Wang and Pessin, 2013).
The miR-29 family comprises miR-29a, miR-29b, and miR-29c. In the cell model of myogenic differentiation, the upregulation of miR-29 can regulate the NF-kB pathway (Wang et al., 2008), TGF-β pathway (Winbanks et al., 2011; Wang et al., 2012), and Akt3 (Wei et al., 2013). In mdx and chronic kidney disease, loss of miR-29 suppresses myogenesis (Wang et al., 2012). MiR-29 also suppresses the myoblasts conversion into myofibroblasts (Wang et al., 2012; Zhou et al., 2012). In muscle progenitor cells, miR-29 can participate in glucose metabolism (Massart et al., 2017), skeletal muscle regeneration (Galimov et al., 2016), and cellular senescence (Hu et al., 2014).
MiR-29 also regulates skeletal muscle protein metabolism. However, conflicting information has been reported. The overexpression of miR-29b has been shown to induce muscle atrophy (Li et al., 2017; Moraes et al., 2017). While the expression of atrophic-related genes is induced in miR-29 knockout mice (Cushing et al., 2011, 2015), and studies have shown that miR-29 attenuates muscle wasting (Silva et al., 2019; Wang et al., 2019). The research is indispensable to uncover the function of miR-29 during skeletal muscle protein synthesis and atrophy. The function of miR-29 can be better explained in vivo, while these studies did not use miR-29 overexpression transgenic mice.
In our study, we found that the disruption of miR-29 led to severe atrophy of skeletal muscle during puberty. The miR-29ab1 cluster overexpression transgenic mice were resistant to denervation-induced and fasting-induced muscle atrophy. Furthermore, the overexpression of miR-29a, b mimics in myotubes could also resist to muscle atrophy. So miR-29a, b function as an inhibitor of the skeletal muscle and myotubes atrophy.
Materials and Methods
Mice
This study was approved by the Institutional Animal Care and Use Committee of China Agricultural University (SKLAB-2015-01-03). All mice, which were raised in specific pathogen-free environments and temperature (25°C ± 1°C), humidity (60–70%), and lighting (a 12 h light and 12 h dark cycle), were regulated. The miR-29ab1 and miR-29b2c double knockout mice (dko) (Ge et al., 2019) and the miR-29ab1 cluster overexpression (dTG) mice were previously reported (Liu et al., 2019). miR-29a/b1 null and miR-29b2/c null mice (Ge et al., 2019) were purchased from Cyagen Biosciences (Beijing, China). TRE-miR-29 mice and ACTA1-rtTA mice (Liu et al., 2019) were purchased from Jifulin Biotech (Beijing, China). Ten double knockout mice (the miR-29ab1 cluster and miR-29b2c cluster dual knockout) and 10 littermates were used. Fifty-four dual transgenic mice (ACTA1-rtTA mice and TRE-miR-29ab1 cluster mice) and 46 littermates were used.
MiR-29ab1 cluster knockout mice and miR-29b2c cluster knockout mice are generated by CRISPR/Cas9 (Ge et al., 2019). To obtain the miR-29ab1 cluster and miR-29b2c cluster dual knockout mice (miR-29ab1−/−:miR-29b2c−/−), we crossed these two types of mice. Finally, dko mice were obtained by crossing miR-29ab1+/−:miR-29b2c−/− mice with miR-29ab1+/−:miR-29b2c+/− mice, or miR-29ab1+/−:miR-29b2c+/− mice with miR-29ab1+/−:miR-29b2c+/− mice. In the absence of wild-type mice among the littermates of dko mice, we chose heterozygous mice (miR-29ab1+/− mice or miR-29b2c+/− mice) as controls. There were no differences in skeletal muscle atrophy-related genes, skeletal muscle weight, and body weight between heterozygous mice and wild-type mice (data not shown).
For dual transgenic mice and littermates, doxycycline (Dox) (2 g/L) and sucrose (10 g/L) were added into the drinking water to induce expression of miR-29ab1 cluster. This concentration of Dox is safe and nontoxic (Urlinger et al., 2000).
Denervation experiments were conducted as previously described (Milan et al., 2015). In brief, mice were anesthetized, the left leg muscles were blunt dissected, and the sciatic nerves were isolated and cut. The right leg underwent a sham operation with blunt separation of muscles but without cutting of the nerve. Mice were sutured after completion.
Fasting experiments were conducted as previously described (Milan et al., 2015). In brief, the control animals were fed ad libitum; the fasting animals were only fed with water.
Histology, immunochemistry, and immunofluorescence
Skeletal muscles were cut, fixed in 4% PFA overnight, and then common paraffin imbedded. The antigen retrieval was in high barometric pressure. Five percent goat serum was used to blocking. Immunostaining with anti-laminin (L9393; Sigma) and anti-myosin (M4276; Sigma) was performed overnight at 4°C (Liu et al., 2015). The sections were incubated with Alexa 488-labeled goat anti-mouse immunoglobulin G/Alexa 488-labeled goat anti-rabbit immunoglobulin G/Alexa 594-labeled goat anti-mouse immunoglobulin G (Invitrogen) (Chen et al., 2017).
Cell culture
C2C12 (Cell Resource Center, Beijing, China) mouse skeletal myoblasts were cultured in DMEM with 10% FBS and 1% penicillin and streptomycin. To induce myogenic differentiation, 2% horse serum in DMEM were used. To induce myotubes atrophy, 10 μM dexamethasone (Dex) were used.
HEK293T (Cell Resource Center) were cultured in DMEM with 10% FBS and 1% penicillin and streptomycin.
C2C12 cells were transfected with 20 nM (myoblasts) or 100 nM (myotubes) concentration of miRNA mimics, miRNA inhibitor, negative control (NC), and inhibitor negative control (INC), which were commercially synthesized from RIBOBIO (Guangzhou, China).
miR-29a mimics forward, 5′-UAGCACCAUCUGAAAUCGGUUA-3′, and reverse, 5′-ACCGAUUUCAGAUGGUGCUAUU-3′;
miR-29b mimics forward, 5′-UAGCACCAUUUGAAAUCAGUGUU-3′, and reverse, 5′-CACUGAUUUCAAAUGGUGCUAUU-3′;
negative control (NC) forward, 5′-UUCUCCGAACGUGUCACGUTT-3′ and reverse, 5′-ACGUGACACGUUCGGAGAATT-3′.
miR-29a inhibitor, 5′-UAACCGAUUUCAGAUGGUGCUA-3′,
miR-29b inhibitor, 5′-AACACUGAUUUCAAAUGGUGCUA-3′,
inhibitor negative control (INC), 5′-CAGUACUUUUGUGUAGUACAA-3′.
Luciferase reporter assay
HEK293T cells were cultured using DMEM containing 10% FBS as well as 1% streptomycin and penicillin. HEK293T cells were seeded to a 24-well plate, 24 h in advance. Luciferase reporter assay was used psiCHECK™-2 vector and the dual luciferase assay system (Promega). psiCHECK-2 vector is a common vector used to detect whether miRNA directly affects the expression of the target gene. Renilla luciferase was used as the main reporter gene, and firefly luciferase was designed for normalizing the expression of renilla luciferase. The 3′UTR of MuRF1 containing the miR-29 target sites or the mutated 3′UTR of MuRF1 without the miR-29 target sites were cloned into the 3′UTR of the firefly luciferase of the psiCHECK-2 vector.
Real-time polymerase chain reaction
Total RNA was isolated using the mirVana™ miRNA Isolation Kit (Life Technologies). The RT primers sequences for miRNA are as follows: miR-29a RT stem-loop primer, CTCAACTGGTGTCGTGGAGTCGGCAATTCAGTTGAGTAACCGAT; miR-29b RT stem-loop primer, CTCAACTGGTGTCGTGGAGTCGGCAATTCAGTTGAGAACACTGA; U6 RT primer, CGCTTCACGAATTTGCGTGTCAT.
For analysis of miRNA expression, the sequences of the primers are as follows:
miR-29a forward 5′-TCACGTAGCACCATCTGAA-3′
miR-29b forward 5′-TCACGTAGCACCATTTGAAA-3′
miR-29a/miR-29b reverse 5′-TCAACTGGTGTCGTGGAGT-3′;
U6 forward 5′-CTCGCTTCGGCAGCACA-3′
U6 reverse 5′-AACGCTTCACGAATTTGCGT-3′.
For analysis of mRNA expression, the sequences of the main primers are as follows:
CTSL, 5′-GTGGACTGTTCTCACGCTCA-3′ and 5′-TGTCATTAGCCACAGCGAAC-3′;
Atrogin-1, 5′-AGCGCTTCTTGGATGAGAAA-3′ and 5′-ACGTCGTAGTTCAGGCTGCT-3′;
MuRF1, 5′-TGGAAACGCT ATGGAGAACC-3′ and 5′-AACGACCTCC AGACATGGAC-3′;
GAPDH, 5′-GGCTGCCCAGAACATCAT-3′ and 5′-CGGACACATTGGGGGTAG-3′.
GAPDH was used as the internal standard.
Western blotting
The following antibodies were used: anti-GAPDH (2118S; Cell Signaling Technology), anti-Fbx32 (3535-100; BioVision), Anti-MuRF1 (ab77577; Abcam), and anti-myosin (M4276; Sigma).
Statistical analysis
Experiments were performed at least three times and the values represent the mean ± SEM. All p-values were calculated using the two-tailed Student t-test with SPSS.
The cross-sectional area was analyzed using ImageJ. For the cross-sectional area and the myotube diameters measurement, first, a line was determined based on the length of the scale, and then this line is set according to the length of the scale. The circumference of the muscle fibers is circled to calculate the cross-sectional area.
Results
Disruption of miR-29 leads to severe skeletal muscle atrophy
To investigate the function of miR-29, we used the miR-29ab1 cluster and miR-29b2c cluster dual knockout mice (dko) generated by CRISPR/Cas9 (Ge et al., 2019). The body weights of the dko mice were significantly lower than those of the controls (Fig. 1A, B). This result is consistent with the results of other studies using miR-29 knockout mice constructed by Cre-floxp system (Cushing et al., 2015; Dooley et al., 2016). To better study the effects of miR-29 on skeletal muscle, we measured the muscle to body weight ratio and found that the ratio in the dko mice was lower than that in the controls (Fig. 1C). The cross-sectional area of the tibialis anterior (TA) muscle fibers was also significantly decreased in the dko mice (Fig. 1D). These results indicate that the loss of miR-29 may cause atrophy of skeletal muscle.
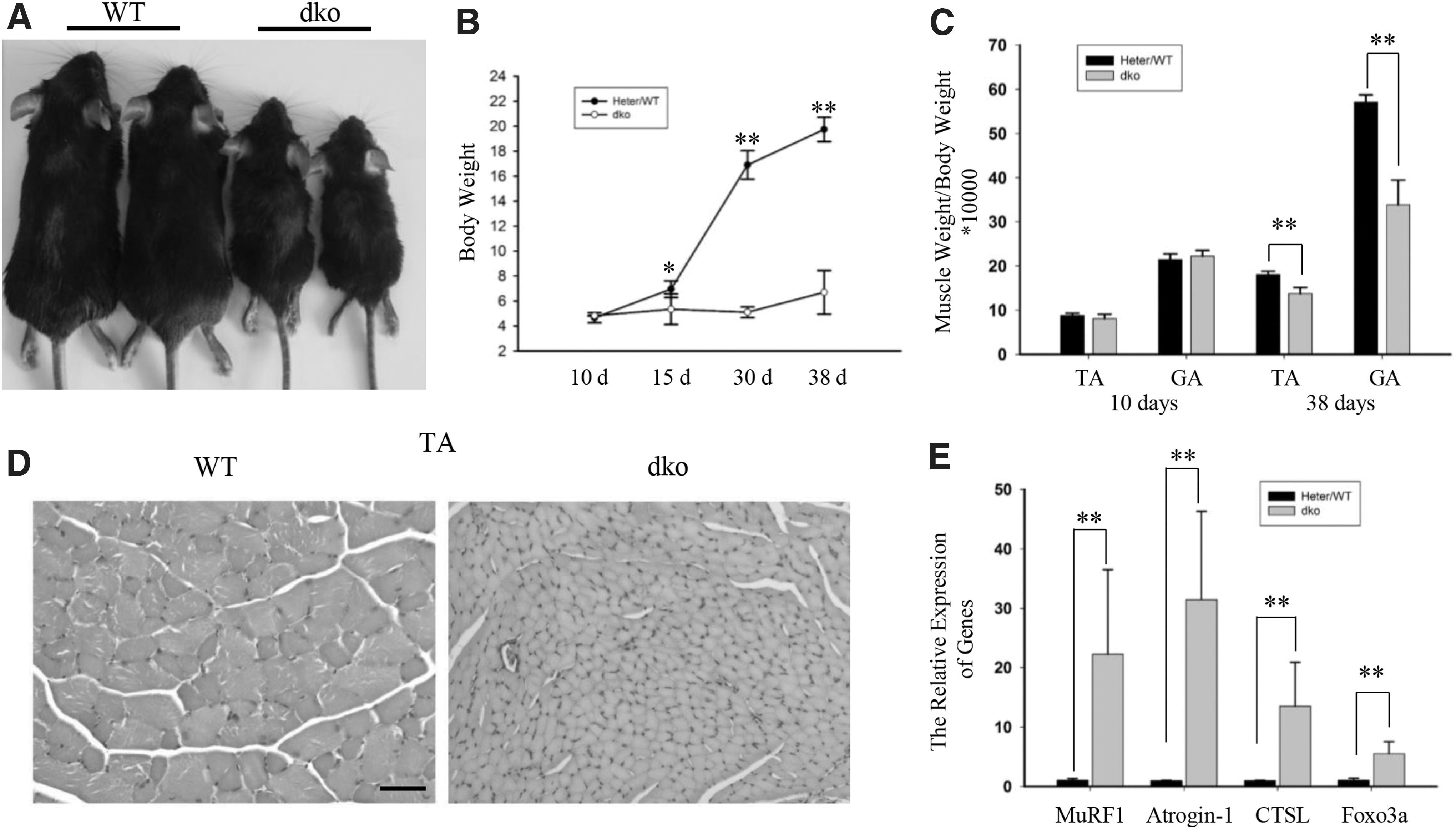
Disruption of miR-29 leads to severe skeletal muscle atrophy.
Some genes can be used as the marker for skeletal muscle atrophy (Lecker et al., 2004; Cohen et al., 2009; Glass, 2010), so we also tested the expression of these atrophy-related genes. Consistent with the skeletal muscle weight, we found that most of these genes are upregulated (Fig. 1E and Supplementary Fig. S1).
Skeletal muscle dysplasia also affects muscle mass (Liu et al., 2019). Also, miR-29 is related to skeletal muscle injury and regeneration (Galimov et al., 2016; Liu et al., 2019). It can be found from Figure 1D that skeletal muscles are not severely damaged. Expression of eMyHC and centrally located nuclei are related to skeletal muscle injury (Relaix and Zammit, 2012). We found no eMyHC-positive myofibers, and centrally located nuclei were observed (Supplementary Fig. S2A). However, real-time polymerase chain reaction (RT-PCR) analysis revealed that the expression of MYH8 and Pax7 was upregulated in the dko mice (Supplementary Fig. S2B). But this change was small for the expression changes of these two genes in dysplasia (Liu et al., 2019). These results suggested that the weight loss of skeletal muscle in miR-29 knockout mice is not mainly due to dysplasia, but due to atrophy.
Overexpression of the miR-29ab1 cluster prevents muscle loss in adulthood
To examine the relationship between miR-29 and skeletal muscle atrophy, we examined the expression levels of miR-29a, b in skeletal muscle atrophy models (denervation or fasting). MiR-29a, b expression was increased in skeletal muscle atrophy (Supplementary Fig. S3). To further test the function of miR-29, we used mice in which miR-29ab1 cluster could be induced in the muscle fibers with temporal specificity (Liu et al., 2019). Starting at 8 weeks of age, we fed mice with Dox for 7, 10, 21, and 28 days, and then quantified weight of TA muscle and gastrocnemius (GA) muscle, the dTG mice were comparable with those in littermates (Fig. 2).
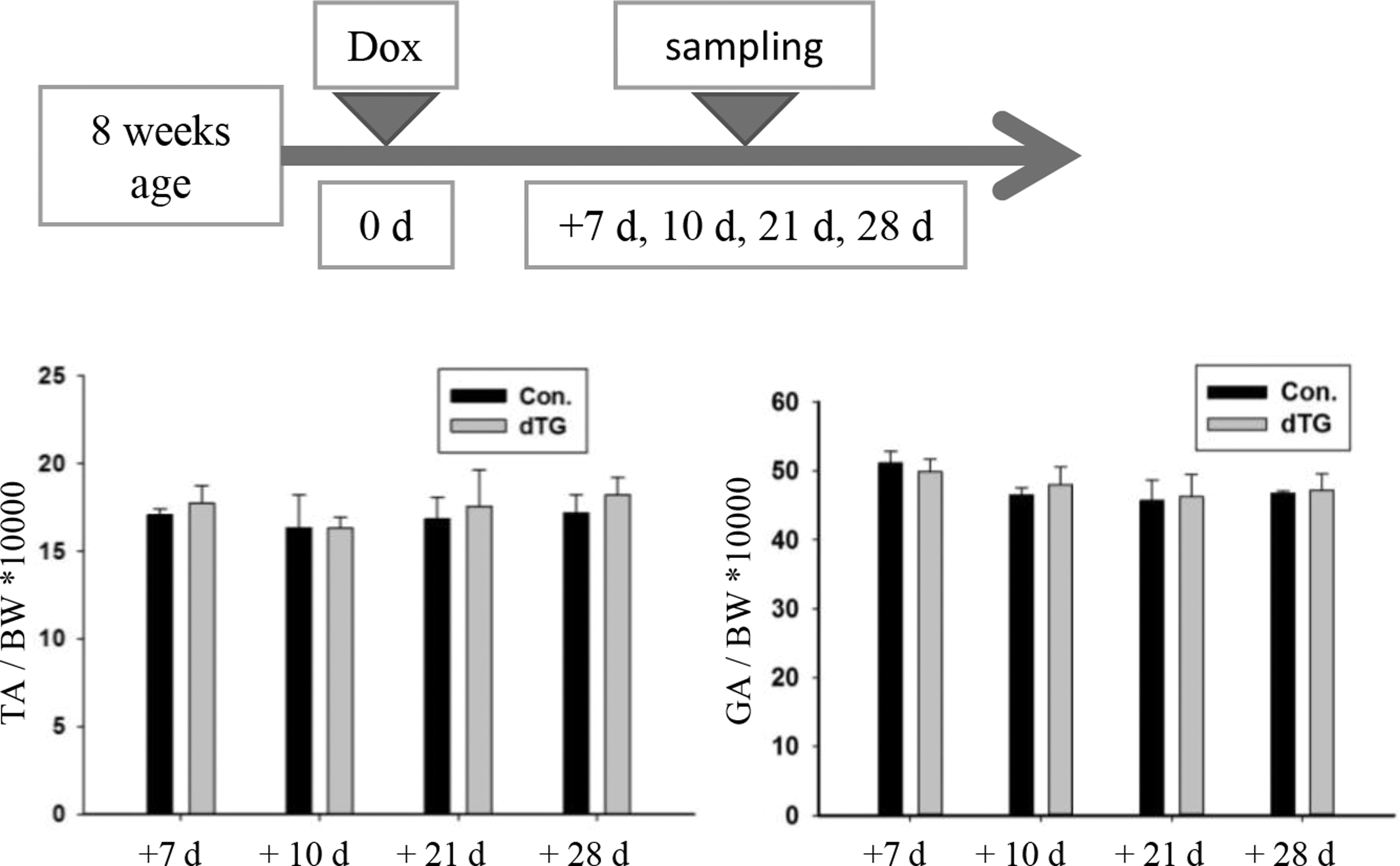
The muscles weight of dTG mice is normal. The muscles weight to BW ratio in the control and dTG mice. n = 5. BW, body weight.
Next, we used denervation as a model of muscle atrophy (Sartori et al., 2013) to investigate the function of miR-29 under the conditions of muscle wasting. In these experiments, the miR-29ab1 cluster was induced in 2-month-old mice, and the mouse sciatic nerve was removed at the time of induction (Fig. 3A). Compared with the controls, the dTG mice exhibited significantly reduced muscle loss after the denervation (Fig. 3B). According to the quantification of the fiber size, the muscles in the dTG mice were partially protected from atrophy (Fig. 3C, D). The expression levels of MuRF1 and Atrogin-1 were dramatically increased during the denervation, and these genes were less upregulated in the dTG atrophied muscle (Fig. 3E). Altogether, miR-29ab1 cluster can regulate the expression levels of the E3 ubiquitin ligases to prevent muscle mass loss.
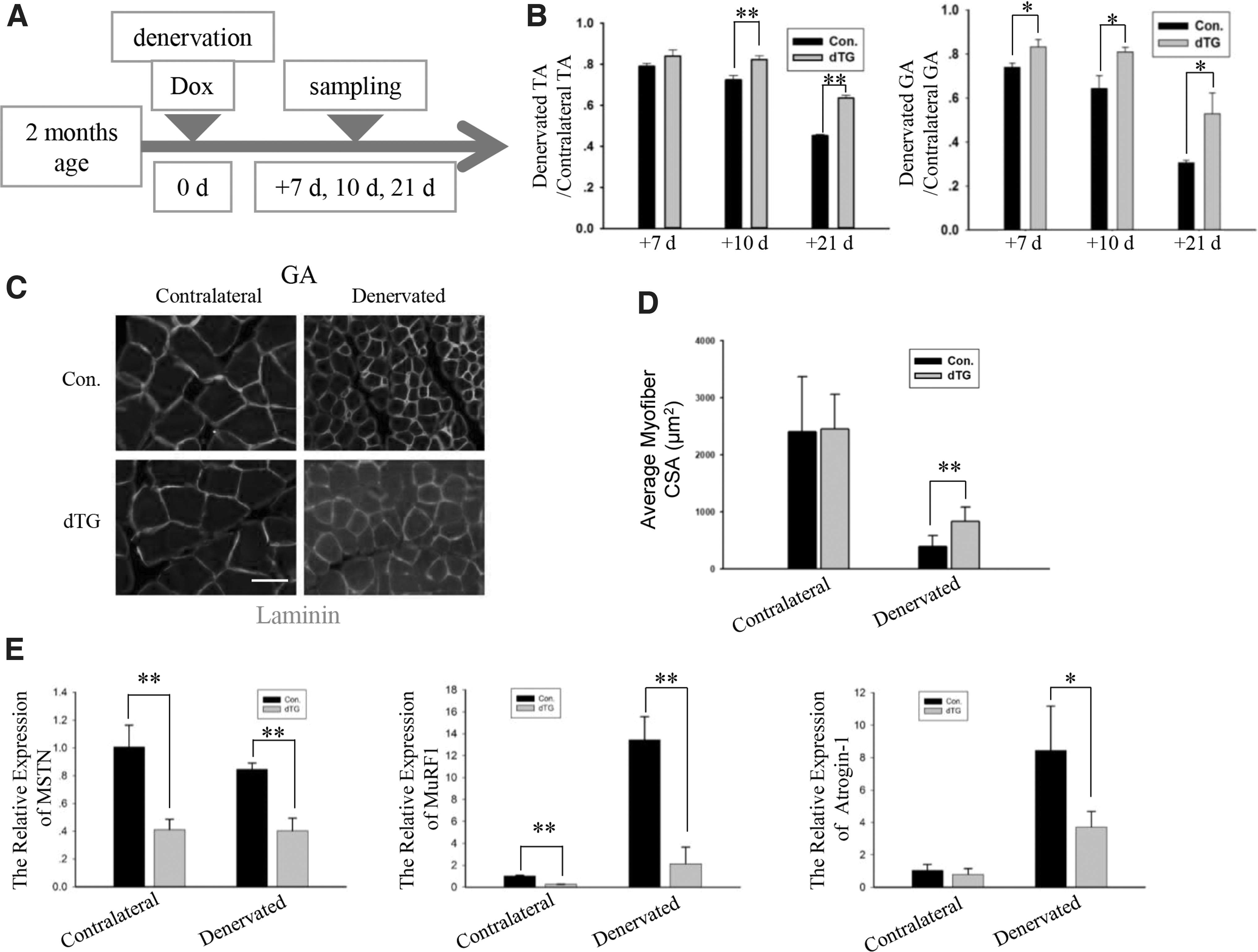
The dTG mice exhibit reduced muscle loss after the denervation in adulthood.
To further determine the function of miR-29 in another model of muscle atrophy, we chose fasting to test the properties of skeletal muscle in the dTG mice (Fig. 4A). In this model, the ratios of the muscles in the fasting dTG mice were significantly higher than those in the fasting controls (Fig. 4B). But for the quantification of fiber size, the dTG mice did not show resistance to muscle wasting (Fig. 4C, D). The expression levels of Atrogin-1 and MuRF1 were lower in the fasting dTG mice than that in the fasting controls (Fig. 4E).

The dTG mice resist fasting-induced muscle atrophy.
MuRF1 is a direct target gene of miR-29a, b
Subsequently, we determined whether miR-29a, b directly inhibit the expression of genes regulating protein degradation. C2C12 cells were transfected with miR-29a, b mimics or miR-29a, b inhibitor (Supplementary Fig. S4). According to the RT-PCR and western blot analyses, the miR-29a, b mimics decreased the expression level of MuRF1, but not of Atrogin-1, in the C2C12 cells (Fig. 5A). Furthermore, the miR-29a, b inhibitor upregulated the expression of MuRF1 (Fig. 5A).
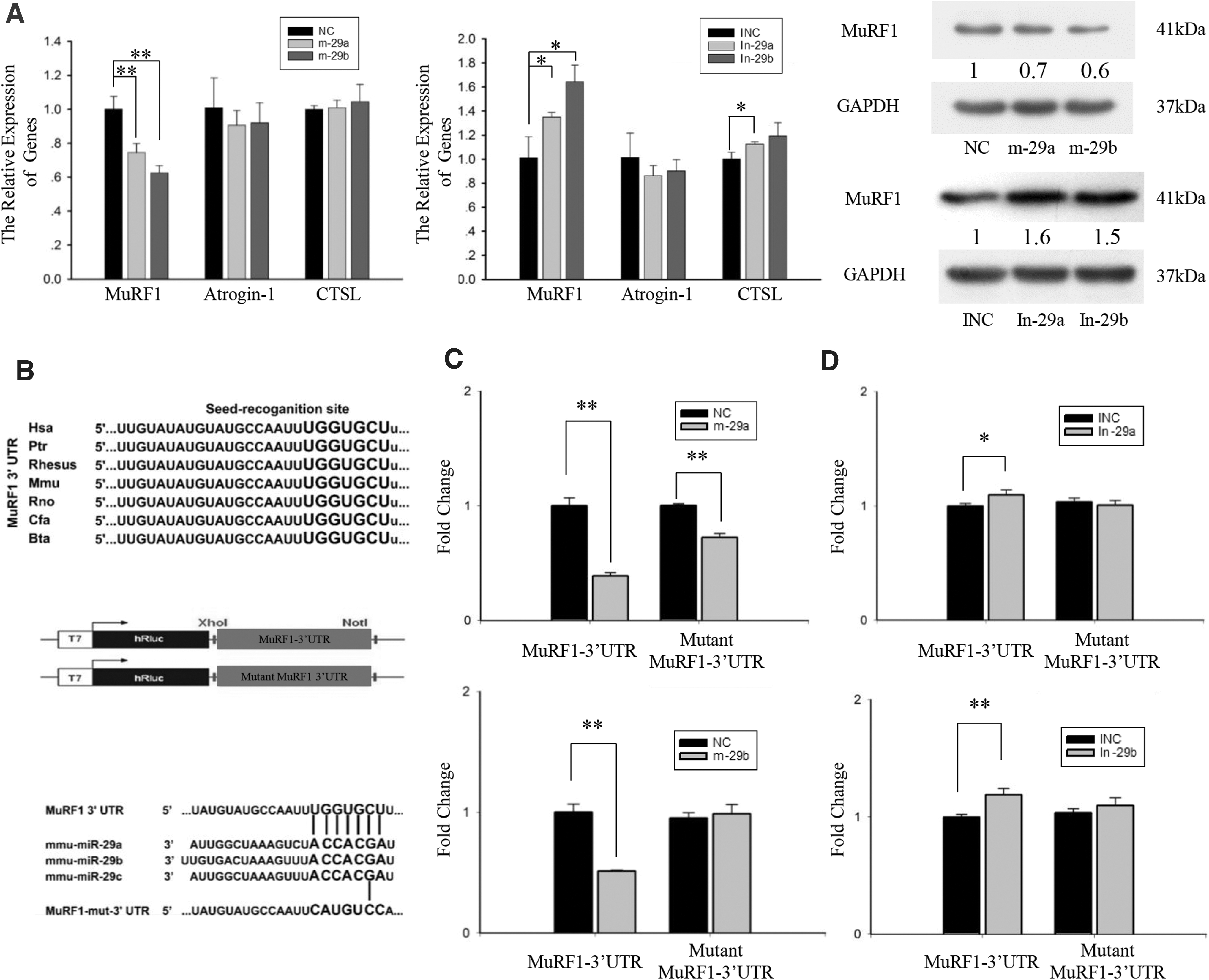
MuRF1 is a direct target gene of miR-29a, b.
Analysis using TargetScan 6.2 showed that the 3′UTRs of MuRF1 contains a conserved complementary sequence of miR-29. In the MuRF1 knockout mice, the expression of Atrogin-1 is also inhibited during skeletal muscle atrophy (Baehr et al., 2011). Therefore, the downregulation of Atrogin-1 in the dTG mice maybe due to the inhibition of MuRF1.
The predicted binding sites of MuRF1 are conserved in several species (Fig. 5B). The putative 3′UTR target site or mutation site was cloned downstream of the reporter gene (Fig. 5B). Subsequently, we cotransfected the psiCHECK-2 vector (wild-type MuRF1 or mutant MuRF1) with miR-29a, b mimics or NC into HEK293T cells. The normalized luciferase activity of cells cotransfected with miR-29a mimics and wild-type MuRF1 vector was reduced 60% (Fig. 5C). When the binding site was mutated, there was some nonspecific inhibition, but only a 25% reduction. The relative luciferase activity in the cells cotransfected with the miR-29b mimics and wild-type MuRF1 vector was significantly lower than that in the cells cotransfected with NC and wild-type MuRF1 vector. No changes were observed in the relative luciferase activity of mutant MuRF1 vector between the miR-29b mimics and NC.
We also cotransfected the psiCHECK-2 vector and miR-29a, b inhibitor or INC into HEK293T cells. The relative luciferase activity in the cells cotransfected with the miR-29a, b inhibitor and wild-type MuRF1 vector was significantly higher than that in the cells cotransfected with the INC and wild-type MuRF1 vector. The relative luciferase activity of mutant MuRF1 vector did not differ between the cells transfected with the miR-29a, b inhibitor and INC (Fig. 5D). Thus, MuRF1 is a direct target gene of miR-29a, b.
MiR-29a, b mimics protect against myotubes atrophy
The abovementioned data prompted us to determine whether miR-29a, b can prevent atrophy at the cellular level. Four-day differentiated myotubes were transfected with miR-29a, b mimics or NC, and then, 10 μM Dex were added to the cells 6 h after the transfection. Thirty-six hours after the miR-29a, b mimics transfection, the myotubes were labeled by immunofluorescence (Fig. 6A and Supplementary Fig. S5). In myotubes transfected with NC, miR-29a mimics, or miR-29b mimics, there was no significant difference in myotube diameters (Supplementary Fig. S5). Myotubes treated with NC and Dex exhibited a significant decrease in diameter, whereas the myotubes treated with the miR-29a, b mimics and Dex showed a less decrease in diameter (Fig. 6B). Expectedly, according to the western blot analysis, the Dex-mediated reduction in myosin was decreased in C2C12 myotubes transfected with the miR-29a, b mimics (Fig. 6C, D).
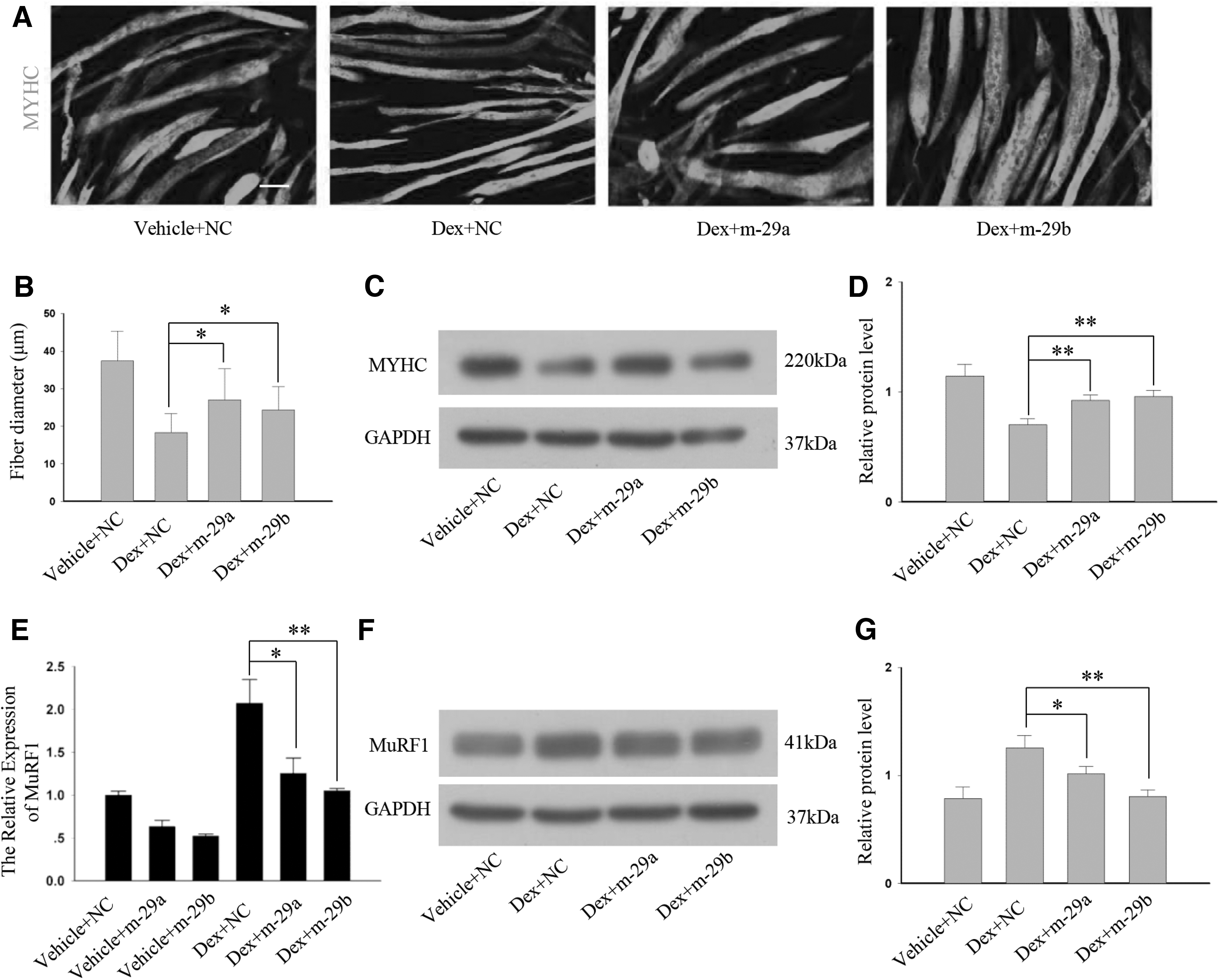
MiR-29a, b mimics prevent myotubes atrophy.
Therefore, the role of the miR-29a, b mimics in Dex-induced myotubes atrophy was confirmed. Furthermore, according to the RT-PCR and western blot analysis, the miR-29a, b mimics decreased the expression level of MuRF1 in the Dex-treated myotubes (Fig. 6E–G). Thus, miR-29a, b mimics can prevent atrophy by regulating the expression of the E3 ubiquitin ligases.
Discussion
MiR-29 family is important in diverse biological processes, including fibrosis (Zhang et al., 2013), myoblasts differentiation (Wang et al., 2008; Winbanks et al., 2011; Wei et al., 2013), and protein balance (Cushing et al., 2015; Li et al., 2017). Our work and that of others have shown that muscle atrophy is regulated by a transcription-dependent process that requires the expression of atrophy-related genes (Lecker et al., 2004; Glass, 2010; Liu et al., 2017). In this study, our findings highlight that miR-29ab1 cluster is resistant to muscular atrophy.
Studies have investigated the regulation of skeletal muscle protein synthesis and atrophy by miR-29. Using miR-29 knockout mice, miR-29 was shown to inhibit the expression of atrophy-related genes (Cushing et al., 2011, 2015). In a subsequent study, overexpression of miR-29b used agomir in vivo, Li et al. (2017) found that the miR-29b agomir induces skeletal muscle atrophy. The miR-29b agomir enters into the skeletal muscle and may cause minor damage to the skeletal muscle, and miR-29b can inhibit the extracellular matrix of skeletal muscle (Liu et al., 2019). Therefore, using miR-29b agomir to detect the function of miR-29b in skeletal muscle may not be suitable.
At the cellular level, the myotubes were transfected with miR-29b mimics. Moraes et al. (2017) found that the miR-29b mimics induce skeletal muscle atrophy. Well, our study showed that miR-29ab1 cluster prevents muscle atrophy using the miR-29ab1 cluster-overexpressing mice. Also, we found that miR-29a, b mimics protect against myotubes atrophy. The differentiated myotubes were very important for this experiment. The heterogeneity of differentiated myotubes of their experiment may be a cause of abnormal experimental results. More studies have indicated that miR-29 resists muscle wasting (Cushing et al., 2011, 2015; Silva et al., 2019; Wang et al., 2019).
Hu et al. (2014) found that miR-29 induces cellular senescence in aging muscle. They studied the function of miR-29 in muscle progenitor cells. Galimov et al. (2016) also use muscle progenitor to study the function of miR-29 in skeletal muscle regeneration. Muscle progenitor cells are different from muscle fibers. Muscle fibers and miR-29 in the muscle fibers should also play important roles in these processes. Related research should be carried out in the future.
MuRF1 is a direct target gene of miR-29a, b. In dTG mice, the miR-29ab1 cluster prevented muscle loss by inhibiting the expression of MuRF1, and the expression level of skeletal muscle atrophy-related genes was also downregulated. We can find that Atrogin-1 was decreased in dTG mice in Figures 3E and 4E. In Figure 5, C2C12 cells were transfected with miRNA mimics. We detected the expression level of related genes in a short time (12 h) after transfection. So, we can find that miR-29a, b mimics inhibited the expression of the target gene, but not of Atrogin-1 in the C2C12 cells.
Atrophy of muscle due to aging touches everyone (Lexell et al., 1988). Therapy for muscle atrophy is important for improving the length and quality of life. Different catabolic stimuli may have various effects on protein degradation pathways (Lecker et al., 2004). In the various in vivo and in vitro atrophy models, the overexpression of miR-29a, b prevented skeletal muscle atrophy.
The disruption of miR-29 led to severe skeletal muscle atrophy. Consistently, the miR-29ab1 cluster overexpression prevented muscle loss in adulthood mice. Well, the miR-29ab1 cluster knockout mice or miR-29b2c cluster knockout mice had no obvious skeletal muscle atrophy during puberty. When miR-29ab1 cluster was knocked out, the expression of miR-29b2c cluster was sufficient to maintain the skeletal muscle mass. The overexpression of miR-29ab1 cluster can increase the expression of miR-29a and miR-29b by >10 times, and prevent muscle atrophy by inhibiting MuRF1 in atrophy models.
The finding that MuRF1 involved in different catabolic conditions that are under miR-29ab1 cluster regulation is an important step toward the understanding of miR-29ab1 cluster function in skeletal muscle atrophy. Our data from induced miR-29ab1 cluster overexpression mice highlight the concept that miR-29ab1 cluster is a therapeutic option for atrophy.
Conclusion
These results highlight the context-dependent effects of miR-29ab1 cluster and demonstrate that miR-29ab1 cluster plays a key role in the maintenance of skeletal muscle. Now in our data, the dTG mice produced positive therapeutic results. As the small nucleotides, miR-29ab1 cluster is an excellent inhibitor for MuRF1, ultimately suggesting that miR-29ab1 cluster is good therapeutic molecule candidate.
Footnotes
Authors' Contributions
C.L. designed and performed experiments, analyzed data, and wrote the article. L.L., L.G., and C.L. helped perform experiments. M.G. and T.W. helped analyze data; K.Z., Y.S., and Y.Z. helped perform cell culture experiments. M.L. and Y.Y. revised the article. B.Z., G.Z., and Q.M. conceived the study, analyzed the data, and wrote the article. All authors contributed to the final version of the article.
Acknowledgment
We appreciate Yunping Dai for his kind help in animal model preparing.
Data Availability Statement
The data and material are available from the corresponding author on reasonable request.
Disclosure Statement
No competing financial interests exist.
Funding Information
This research was supported by National Natural Science Foundation of China (31790412, 31970712), National Basic Research Program of China (2015CB943103), National Transgenic Breeding Project of China (2016ZX08010004), China Agriculture Research System of MOF and MARA (CARS-36), the Project for Extramural Scientists of State Key Laboratory of Agrobiotechnology (2020SKLAB6-9), and the Natural Science Foundation of Inner Mongolia (2019LH03023), Research Program of Science and Technology at Universities of Inner Mongolia Autonomous Region of China (NJZY20096).
Supplementary Material
Supplementary Figure S1
Supplementary Figure S2
Supplementary Figure S3
Supplementary Figure S4
Supplementary Figure S5
References
Supplementary Material
Please find the following supplemental material available below.
For Open Access articles published under a Creative Commons License, all supplemental material carries the same license as the article it is associated with.
For non-Open Access articles published, all supplemental material carries a non-exclusive license, and permission requests for re-use of supplemental material or any part of supplemental material shall be sent directly to the copyright owner as specified in the copyright notice associated with the article.