
Abstract
Succinate is an important intermediate product of mitochondrial energy metabolism. Recent studies revealed that beyond its known traditional metabolic functions, succinate plays important roles in signal transduction, immunity, inflammation, and posttranslational modification. Recent studies showed that patients and mouse models with cardiovascular disease have high levels of serum succinate and succinate accumulation. Atherosclerosis (As) is the pathological basis of cardiovascular and peripheral vascular diseases, such as coronary heart disease, cerebral infarction, and peripheral vascular disease, and is a major factor affecting human health. This article reviews the progression of succinate in As diseases and its underlying mechanisms.
Introduction
Cardiovascular disease is the leading cause of death worldwide. Atherosclerosis (As), the common pathological basis of cardiovascular and cerebrovascular diseases (Beverly and Budoff, 2020), starts with the formation of fat streaks on the intima, which then develop into fibrous and atheromatous plaques under the stimulation of pro-As risk factors (Badimon and Vilahur, 2014). Endothelial cells (ECs) are activated by mechanical or chemical stimulation, and their dysfunction is the key step of the initiation of As. Activated ECs synthesize growth factors and chemokines, which contribute to the vascular smooth muscle cell (SMC) phenotype transformation, leading to the progression of atherosclerotic lesions and the formation of fibrous plaques.
In addition, macrophages and dendritic cells (DCs) are recruited and adhere onto activated ECs and invade the intima. The dynamic interaction and crosstalk of these cells enhance the progression of atherosclerotic lesions and contribute to the instability (Tabas et al., 2015; Gimbrone and Garcia-Cardena, 2016). When unstable plaques rupture, thrombus components are released into the blood, leading to serious clinical events, such as stroke, myocardial infarction, and even death (Foks and Bot, 2017).
Recent studies have showed that the serum succinate content in the hyperlipidemia, and As mouse models are remarkably increased, and excess succinate participates in multiple pathological processes of atherosclerotic lesions (Yang et al., 2016; Zhang et al., 2018; Martinez-Reyes and Chandel, 2020). In this review, existing evidence and the role of succinate in atherosclerotic cardiovascular disease are summarized and discussed.
Source and Function of Succinate
Succinate, also known as butanedioic acid, is generated in the mitochondrial matrix during the tricarboxylic acid cycle (TCA) and catalyzed by succinyl-CoA synthetase coupling with the production of high-energy nucleoside triphosphate (Ryan and O'Neill, 2017). Succinate is oxidized to fumarate by succinate dehydrogenase (SDH), which process is reversible and releases reactive oxygen species (ROS) (Ryan et al., 2019).
The release of succinate from mitochondria into the cytoplasm is mediated by mitochondrial dicarboxylate carriers and voltage-dependent anion-selective channel (Hodge et al., 1997). Excess intracellular succinate is transferred to the outside of the cell through organic anion/dicarboxylic acid transporter and monocarboxylic acid transporter in a pH-dependent manner (Reddy et al., 2020). In addition, extracellular succinate can be reabsorbed by cells through sodium-dependent dicarboxylic acid transporters.
Under physiological conditions, the concentration of succinate in the blood is approximately micromolar (Peti-Peterdi, 2010; Hu et al., 2017). However, high glucose, high fat, hypoxia, and lipopolysaccharide (LPS) can induce intracellular succinate accumulation and can increase to millimolar concentrations in neuroblastoma cells and VM-M3 cancer cells (Laukka et al., 2016; Flores et al., 2018). The accumulation of succinate is related to the following factors. (1) The activity or function of SDH is impaired, which results in the accumulation of succinate as a reaction substrate. (2) The increased glutamine by entering α-ketoglutarate into the TCA cycle or “γ-aminobutyric acid shunt” is metabolized to produce succinate (Jiang and Yan, 2017; Ryan et al., 2019). (3) The glyoxylate shunt converts isocitrate into succinate (Quartararo et al., 2013).
Physiological succinate can participate in the energy metabolism to produce adenosine triphosphate (ATP), but accumulated succinate can have pathological effects. Wu et al. showed that succinate promotes neuron mitochondrial integrity and ischemic stroke through dynamin-related protein 1 (Drp1)-mediated mitochondrial fission (Wu et al., 2017). During ischemia–reperfusion, succinate accumulates in hypoxic cardiomyocytes, further aggravating myocardial damage (Zhang et al., 2018).
In patients with lung cancer, the elevated serum succinate promotes tumor metastasis and has emerged as a biomarker (Wu et al., 2020). Intracellular succinate interferes with energy metabolism via the hypoxia-inducible factor 1-alpha (HIF-1α) and then activates its specific receptor SUCNR1, belonging to the G protein-coupled receptor (GPR) family and also named as GPR91, which aggravates synovial inflammation and angiogenesis (Sadagopan et al., 2007).
Extracellular succinate contributes to the development of normal retinal ischemic retinopathy, and the pathogenesis of proliferative diabetic nephropathy through the activation of SUCNR1 (Hu et al., 2017; Chen et al., 2019). Lu et al. (2018) showed that extracellular succinate enhanced localization of mitochondrial fission factor and Drp1 in mitochondria promoted mitochondrial fission, leading to mitochondrial dysfunction and cardiomyocyte apoptosis.
Several pathways explain the pathological roles of excess succinate: (1) upregulation of mitochondrial ROS (Mills and O'Neill, 2014); (2) direct reversal of the GTP produced with the action of succinate-CoA ligase at the substrate phosphorylation level and inhibition of the use of ketone bodies in the brain when starving (Tretter et al., 2016); (3) inhibition of the prolyl hydroxylase domain (PHD) in the ROS-dependent manner and upregulation of the interleukin (IL)-1β by stabilizing HIF-1α and acting as an inflammation signal (Tannahill et al., 2013); and (4) binding to the membrane SUCNR1 receptor via the solute carrier located in membrane, thereby initiating complex signal transduction (Keiran et al., 2019).
Recently, studies showed that succinate contributes to protein succinylation. This posttranslation modification occurs on lysine residues, resulting in the net charge of the modified lysine residue from +1 to −1, which impacts modified protein function and plays important roles in a variety of life activities and diseases (Ali et al., 2020). Xiao et al. (2021) showed that the disruption of succinylation is a biochemical hallmark of subarachnoid hemorrhage in mice and the intervention of succinylation is a promising therapeutic strategy. Liu et al. (2021) showed that during pregnancy, dietary succinate supplementation induces histone succinylation in the Ppargc1a promoter, leading the activation of Ppargc1a gene transcription in mice.
Excess Succinate Promotes ECs Dysfunction
Vascular ECs lie on the lumen side of the blood vessel wall, thereby regulating vasomotor blood vessels and maintaining the dynamic balance of anti- and procoagulation (Gimbrone and Garcia-Cardena, 2016). Besides, vascular ECs contribute to immune response and other physiological functions by synthesizing and secreting a variety of cytokines, including angiotensin II (Ang-II), endothelin-1, thromboxane A2, prostacyclin H2, and nitric oxide through autocrine, paracrine, and endocrine pathways. Endothelial dysfunction is characterized with reduced vasodilation, proinflammatory state, and prothrombic properties, which induce adverse biological effects (such as As, coronary artery disease, and hypertension) and participate in the process of their occurrence and development.
Oxidative stress is caused by the unbalanced elimination and production of oxygen free radicals, which can induce the damage of VEC, and is a major factor in atherosclerotic lesions. The energy source of ECs mainly relies on glycolysis, which reduces the consumption of oxygen and the damage to the endothelium by ROS produced by mitochondria.
Ko et al. (2017) showed that succinate increases the fission of mitochondria and the level of mitochondrial ROS in human mesenchymal stem cell, which is mediated by the translocation of cytosolic Drp1 to the mitochondria outer membrane via PKCζ/p38 MAPK pathway. Besides, the oxidation of succinate drives the reverse electron transfer to increase the generation of ROS in complex I, leading to the endothelial protective factor nitric oxide inactivation and endothelial dysfunction (Ryan et al., 2019). Excessive ROS uncouples endothelial nitric oxide synthase to reduce nitric oxide production, thereby aggravating endothelial disorders (Rajendran et al., 2013).
The renin–angiotensin system (RAS) plays key roles in EC homeostasis. Renin is the initiator of RAS, and Ang-II is the main biologically active peptide in RAS. Studies showed that succinate/SUCNR1 promotes the release of renin from the macula densa cell by activating ERK1/2, P38, and COX-2, whereas the secretion of renin is reduced in SUCNR1 knockout mice (Rajendran et al., 2013). Peti showed that the intravenous injection of succinic acid induces mouse hypertension through renin and Ang-II. This effect is not observed in SUCNR1-deficient mice (Peti-Peterdi, 2010).
Amraei and Rahimi N found that Ang-II promotes vasoconstriction through the activation of AT1R and inhibition of the nitric oxide signaling pathway, thereby inducing endothelial dysfunction (Amraei and Rahimi, 2020). The vascular endothelial damage caused by Ang-II can be improved by adiponectin and melatonin through antioxidant and inhibiting apoptosis (Nakao et al., 2013; Zhi et al., 2014). These results indicate that excessive succinate activates the SUCNR1/RAS pathway to promote the secretion of Ang-II, leading to endothelial damage and dysfunction.
SUCNR1 is highly expressed in human platelets. The activation of SUCNR1 promotes the production of thromboxane (TX) A2,12-hydroxy-eicosatetraenoic acid, and 12-hydroxy-heptadecatrienoic acid in human platelets, leading platelet aggregation/activation (Tang et al., 2020). It is now established that platelets contribute to the initiation and progression of atherosclerotic lesions. Activated platelets adhere to the damaged endothelium, aggravating EC injury and inducing inflammatory response (Garshick et al., 2020). Upon the atherosclerotic plaque ruptured, platelets are abnormally activated to form platelets-rich blood clots, which promote the formation of thrombus and results in thrombotic events.
Spath et al. (2012) showed that under low concentrations of exogenous ADP, platelet aggregation is enhanced by succinate with concentration-dependent manner (10–1000 μM). Human platelet aggregation induced by succinate is characterized with increased expression of activated glycoprotein IIb-IIIa and P-selectin, which depended on P2Y(12) activation, thromboxane A(2) generation, and ATP release. This process can be completely prevented by 2Y(12) receptor inhibitor (Högberg et al., 2011).
Overall, the excess succinate reduces the protective effect of endothelium-derived vasoactive substances and increases the endothelial damage mediated by oxidative stress, leading to endothelial dysfunction and the occurrence and development of atherosclerotic lesions (Fig. 1).

The accumulation of succinate promotes the production of reactive oxygen species, which uncouples endothelial nitric oxide synthase and damages the vascular endothelium. Besides, succinate activates the RAS system, glycoprotein (GP) IIb–IIIa, and P-selectin through the receptor SUCNR1 to promote endothelial dysfunction and the formation of thrombosis. RAS, renin–angiotensin system.
Succinate Accumulation Boosts SMC Phenotypic Transition
SMCs are a group of cells with high plasticity located in the middle layer of blood vessels (Basatemur et al., 2019). In accordance with their ability of proliferation, migration, and secretion, SMCs are divided into contractile phenotype and synthetic phenotype. The contractile phenotype has lower proliferation, migration, and secretion than synthetic type (Allahverdian et al., 2018). Under pathological conditions, SMCs in mature blood vessels can undergo phenotypic transition from the contraction state to synthetic proliferating cells (Stegemann et al., 2005).
In the early stage of atherosclerotic lesions, highly proliferative synthetic phenotype SMCs migrate to the intima, producing a large amount of extracellular matrix (ECM) and inflammation factors and promoting the progression of atherosclerotic lesions (Shankman et al., 2015). In the late stage of atherosclerotic lesions, the fibrous cap constructed with SMCs and their ECM can stabilize plaques and inhibit their rupture. SMCs play vital roles in the progression of atherosclerotic lesions and their stability.
Studies have shown that succinate regulates osteoclastogenesis via the nuclear factor kappa beta (NF-κB) signaling in mesenchymal stem cells. The NF-κB signaling is enhanced by succinate through the receptor activator of the NF-κB ligand in RAW 264.7 cells (Guo et al., 2017). In nicotine-induced cardiovascular diseases, nicotine induces the SMC cytoskeleton protein upregulation and migration achieved by the NF-κB signaling pathway (Wang et al., 2013).
The platelet-derived growth factor-BB (PDGF-BB) induces vascular SMC differentiation, proliferation, and migration through the NF-κB/mTOR signaling pathway, which is identified by the increased expression of the synthetic gene osteopontin and the decreased expression of contraction genes in vascular SMC (Lu et al., 2018). In addition, the inhibition of NF-κB can weaken the hydrolysis of matrix metalloproteinase (MMP)-9 and MMP-2 on the ECM in vascular SMC (Eagleton et al., 2006; Guo et al., 2008), which inhibit vascular SMC migration and protect the late As plaque rupture.
Under normal oxygen conditions, HIF-1α is hydroxylated by PHD and degraded by the ubiquitin–protease hydrolysis complex. Studies showed that the accumulation of succinate leads to the accumulation of HIF-1α and the enhancement of its transcriptional activity (Selak et al., 2005). This phenomenon is possible due to Fe2+ (PHD cofactor) oxidized by the oxidative metabolism of succinate. Moreover, HIF-1α promotes the proliferation of human vascular SMCs induced by growth factor, such as fibroblast growth factor 2, PDGF, and epidermal growth factor. Besides, HIF-1α promotes Drp-1-mediated mitochondrial division to increase the proliferation of vascular SMCs in human pulmonary hypertension.
The Drp1 inhibitor Mdivi1 inhibits mitochondrial division, prevents the upregulation of the proliferating cell nuclear antigen and cyclin D1, triggers cell cycle disorders, and decreases HIF-1α expression, thereby inhibiting the vascular SMC proliferation. These results suggest that succinate may enhance the accumulation of HIF-1α to promote SMC proliferation via growth factors and mitochondrial pathways (Marsboom et al., 2012).
Studies found that succinate/SUCNR1 activates the RAS system (Komers, 2013) The released Ang-II induces vascular fibrosis by directly activating the SMAD pathway of SMCs and binding to its specific receptor AT1R (Rodriguez-Vita et al., 2005). Mondaca-Ruff et al. (2018) found that Ang II-induced autophagy with AT1R-dependent mechanism contributes to hypertrophy and proliferation in vascular SMCs. Moreover, Ang-II exerts a positive regulatory effect through nucleolin and regulates the expression and secretion of various growth factors and cytokines to enhance the phenotypic transformation of vascular SMCs (Fang et al., 2020).
In summary, the biological effects mediated by Ang-II release, NF-κB regulation of gene transcription, and HIF-1α stabilization induced by succinate accumulation promote the proliferation and migration of vascular SMCs (Fig. 2), thereby negatively regulating the stability of AS plaques and promoting As.
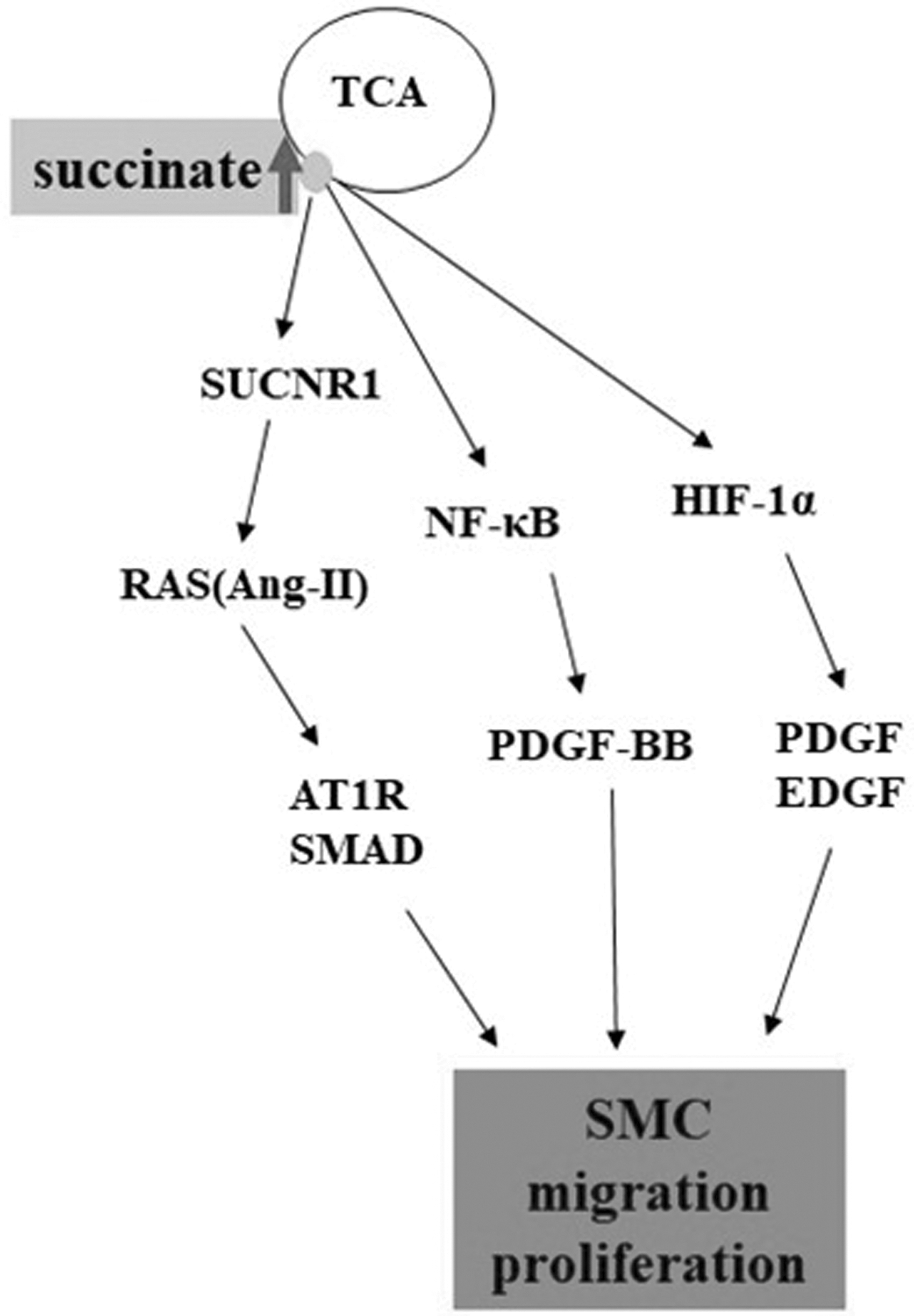
The accumulation of succinate activates the NF-κB, HIF-1α signaling pathway, and the RAS system to induce SMC differentiation, proliferation, migration, and vascular fibrosis. SMC, smooth muscle cell; NF-κB, nuclear factor kappa beta; HIF-1α, hypoxia-inducible factor 1-alpha.
Succinate induces macrophage polarization
Macrophages are important players in the progression of atherosclerotic plaques and determinants of their stability through the accumulation of lipids and the release of proinflammatory cytokines. According to their activation and immune function, macrophages can divide into the proinflammatory phenotype M1 and the anti-inflammatory phenotype M2 (Lee et al., 2018). M1 macrophages express high levels of interleukin-12 (IL-12) and IL-23 and low level of IL-10; secrete inflammatory cytokines, such as tumor necrosis factor (TNF)-α and IL-1β; and participate in antigen presentation. Conversely, M2 macrophages express a high level of IL-10, transforming growth factor-β, and other anti-inflammatory factors, thereby inhibiting inflammatory response and promoting angiogenesis and tissue repair (Viola et al., 2019).
Macrophages can undergo phenotypic transformation response to different microenvironmental stimulations, and this phenomenon is known as macrophage polarization. In atherosclerotic plaque tissues, the macrophage polarization contributes to the progression and stability of atherosclerotic lesions. M1 macrophages enhance inflammation and the accumulation of lipids, which in turn accelerates the transformation of monocyte to macrophage and the formation of foam cell, promoting the progression and enlargement of atherosclerotic plaques. M2 macrophages inhibit plaque progression via anti-inflammatory effects. During the plaque progression stage, M2 macrophages are transformed into M1 macrophages (Chinetti-Gbaguidi et al., 2015).
Interferon (IFN)-γ and LPS cause macrophage energy metabolism reprogramming, which is characterized with the generation of ATP shift from oxidative phosphorylation to glycolysis and the accumulation of succinate. This phenomenon achieves the macrophage polarization to a proinflammatory phenotype (Bories and Leitinger, 2017). Studies have found that the accumulation of succinate in macrophages can directly inhibit the activity of PHD and promote the secretion of IL-1β by increasing the expression of HIF-1α. Shio et al. (2015) found that the use of HIF-1α inhibitors to target downregulation of HIF-1α levels can reduce the production of cytokine IL-1β. It reminds us existing the possibility of succinate promote inflammation via PHD/HIF/IL-1β axis.
Moreover, the maturation and release of IL-1β depend on the activation of the Nod-like receptor 3 (NLRP3) inflammasome. NLRP3 maintains proinflammatory macrophages through the ASC/caspase-1 pathway induced by propofol (Sun et al., 2019). In another in vitro experiment, it was found that cinnamaldehyde can reduce the inflammation induced by succinate accumulation by inhibiting NLRP3 in RAW246.7 cells and rats, which is achieved by suppressing IL-1β through modulating succinate/HIF-1α axis and inhibition of NLRP3 (Liu et al., 2020).
In addition, the oxidation of succinate in the mitochondria enhances the formation of mitochondrial ROS. Yuan et al. (2019) showed that mitochondrial ROS drives M1 macrophage polarization by impairing the autophagy–lysosome system. Clearing ROS can reduce the activation of NLRP3 inflammasome and the release of proinflammatory cytokines (Shio et al., 2015). Besides, ROS can oxidize Fe2+, which can stabilize the HIF-1α to further promote the generation of IL-1β. Both show a trend toward M1-type polarization (Mills et al., 2016).
SUCNR1 is highly expressed on DCs. Succinate affects the production of inflammatory cytokines in DC by activating the receptor SUCNR1 (Saraiva et al., 2018). Succinate cooperates with the Toll-like receptor (TLR) 3 ligand poly(I:C) and TLR7 ligand imiquimod to promote the expression of TNF-α. When DCs are treated with succinate and antigen, the levels of TNF-α and IFN-γ increase. This phenomenon is deprived in SUCNR1-deficient DCs. The proinflammatory cytokines secreted by DCs maintain the M1 polarization of macrophages (Rubic et al., 2008).
These results indicate that succinate promotes the M1 polarization of macrophages through proinflammatory cytokines (Fig. 3). This phenomenon leads to the development of atherosclerotic lesions, which is not conducive to the stability of atherosclerotic plaques.
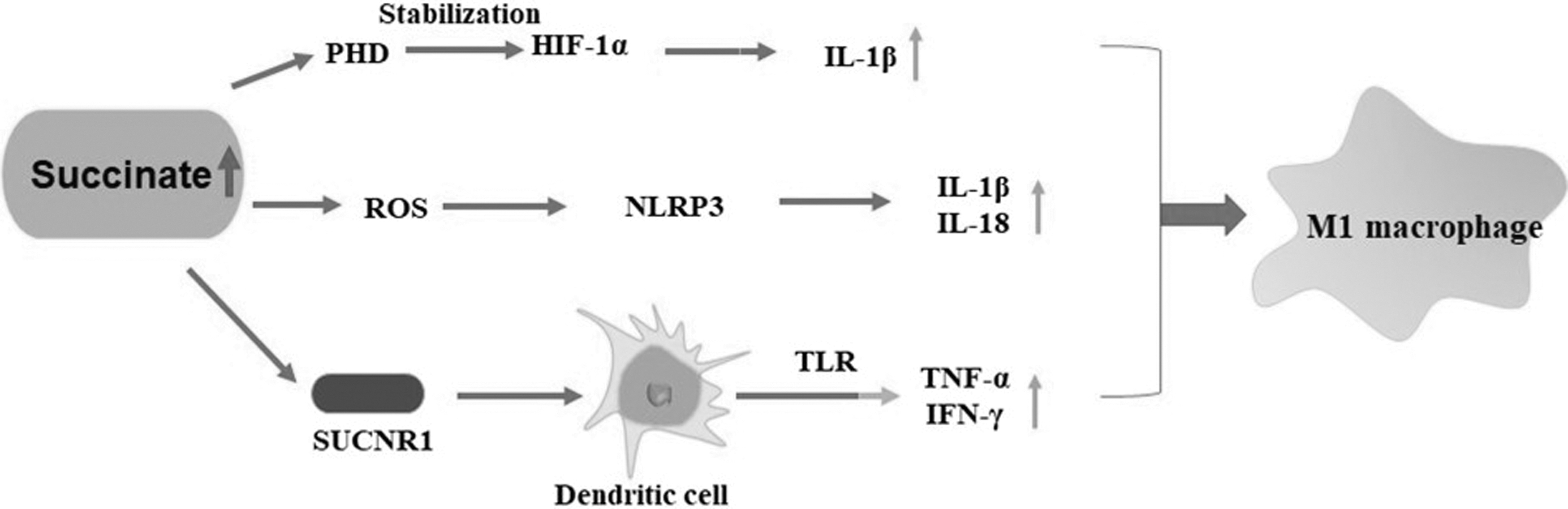
The accumulated succinate inhibits the hydrolysis of PHD and activates the receptor SUCNR1 and the NLRP3 inflammasome to promote the M1 polarization of macrophages. PHD, prolyl hydroxylase domain.
Conclusions and Perspectives
Overall, the excess succinate may be a novel mediator to promote atherosclerotic lesion progression, thereby activating the SUCNR1 signal to induce EC dysfunction and SMC phenotype transformation and macrophage polarization (Fig. 2).
More attention is needed to understand deeply the effect and mechanisms of succinate on atherosclerotic lesions to improve the treatment of As and its complications. GPR146, a member of the GPR family, is recently found to be an effective molecular strategy to reduce plasma cholesterol levels and As (Yu et al., 2019). The activation of SUCNR1 by elevated succinate is involved in inflammatory disease such as intestinal inflammation, joint inflammation, vascular inflammation, and nonalcoholic fatty liver disease. Sucnr1−/− mice were protected against intestinal inflammation and fibrosis induced by the heterotopic transplant of colonic tissue (Macias-Ceja et al., 2019).
Li et al. (2016) showed that Sucnr1 knockdown improved the steatosis in methionine- and choline-deficient diet-fed mice. Therefore, the specific receptor SUCNR1 may be an interesting therapeutic target. In addition, inhibiting the oxidation of succinate to reduce oxidative stress may provide a new direction to inhibit the progress of atherosclerotic lesions and cardiovascular diseases.
Footnotes
Disclosure Statement
No competing financial interests exist.
Funding Information
This work was supported by the grants from the National Natural Science Foundation of China (31870937, 82000421) and the Natural Science Foundation of Hunan Province (2020JJ4076).